 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bin Zhang | + 2541 word(s) | 2541 | 2021-04-21 05:09:45 | | | |
| 2 | Catherine Yang | Meta information modification | 2541 | 2021-04-22 03:36:10 | | |
Video Upload Options
Eukaryotic elongation factor 2 kinase (eEF2K or Ca2+/calmodulin-dependent protein kinase, CAMKIII) is a new member of an atypical α-kinase family different from conventional protein kinases that is now considered as a potential target for the treatment of cancer.
1. Introduction
Targeted therapy is an important strategy for cancer treatment, and this has been well applied in actual clinical applications [1]. At present, most clinically used targeted cancer drugs are inhibitors of tyrosine kinases [1][2]. However, even when these drugs have exceptional efficacy initially, the later emergence of drug resistance limits their usefulness [2]. Finding new drug targets and developing new targeted anticancer agents have accordingly become important aspects of drug discovery and development.
Eukaryotic elongation factor 2 kinase (eEF2K) is the first α-kinase to have been discovered [3]. The activity of eEF2K depends on calcium ions and calmodulin (CaM) [4]. Therefore, eEF2K has also been called CaM-dependent protein kinase III or CAMKIII, and it appears this way in some literature reports [5]. The “α-kinase” is an atypical protein kinase family, and while these often have similar ATP binding pockets to typical protein kinase family members, they are differentiated by having a translocated conserved region with an alternate sequence, GXGXXG [6]. Because of this, drugs that target eEF2K are less likely to modulate typical kinases and should ideally not have or create cross-resistance with traditional kinase drugs. Increasing evidence shows that eEF2K is highly expressed in a variety of tumor tissues and that it is related to the development and prognosis of several kinds of malignancies such as breast cancer, ovarian cancer, colon cancer, glioma, medulloblastoma, hepatocellular carcinoma, and prostate cancer [7][8][9][10][11][12]. In addition, eEF2K can also participate in the regulation of the tumor cell cycle, proliferation, autophagy, apoptosis, angiogenesis, invasion, and metastasis, among other processes [13][14][15]. For all of these reasons, eEF2K is a potential therapeutic target for anticancer drug development.
2. The Role of eEF2K in Cancer
It has been shown that eEF2K is overexpressed and regulates tumor progression in several types of malignancies, including breast cancer, glioma cancer, pancreatic cancer, lung cancer, neuroblastoma, and colorectal cancer [8][16][17]. Previous studies have found that eEF2K is associated with tumor proliferation and survival, tumorigenesis, invasion, drug resistance, and poor prognosis [18][19]. For instance, microRNA 603 inhibits tumor formation in triple-negative breast cancer by the targeted inhibition of eEF2K [20]. It was also reported that eEF2K promotes the proliferation of ovarian cancer cells and that its expression is positively correlated with poor prognosis [19]. In hepatocellular carcinoma, eEF2K promotes angiogenesis through PI3K/Akt and STAT3 signaling [11]. Similarly, eEF2K is positively correlated with lung cancer proliferation, invasion and metastasis, and poor prognosis [21]. These early data indicate the importance of eEF2K in cancers, and suggest that it is a potential new target for cancer chemotherapeutic treatments.
2.1. eEF2K Helps Tumor Cells to Cope with Harsh Environments
The rapid proliferation of cancer cells in tumors requires a high amount of energy, in part due to greatly upregulated protein synthesis. The process leads to tumors creating their own harsh microenvironments that have, for example, reduced availability of nutrients, low pH, and insufficient oxygen. Further adaptation of the tumor cells, including by increasing expression of eEF2K, helps to regulate the synthesis of proteins in the harsh environment and be protected for continued proliferation. Under conditions of nutritional deprivation, tumor cells with high eEF2K expression can continue to survive, while those with low eEF2K expression have been shown to die [22]. When intracellular nutrition is insufficient, the content of ATP decreases, and AMP or ADP increases to activate AMP-activated protein kinase (AMPK). After activation, AMPK can induce the phosphorylation of eEF2K at Ser398 or Ser491 to thereby inhibit the function of eEF2 (Figure 1). This ultimately reduces the rate and energy consumption of intracellular proliferation and protein synthesis and promotes energy production processes such as glucose metabolism and fatty acid oxidation. The mammalian target of rapamycin complex 1 (mTORC1) is another energy-related protein that has been found to downregulate eEF2K [23]. Activated mTORC1 inhibits the activation of eEF2K by inducing its phosphorylation at a variety of residues, including Ser70, Ser78, Ser359, Ser366, Ser392, Ser396, and Ser470 [24][25]. Regarding its regulation, mTORC1 is stimulated by amino acids, hormones, growth factors, and cellular nutrients [4][23]. Under nutritional deprivation, the activity of mTORC1 is inhibited, partially alleviating its phosphorylation and inhibitory effect on eEF2K. At the same time, activated AMPK has been shown to further indirectly inhibit the activity of mTORC1 [26]. In addition, mTORC1-mediated inhibition of eEF2K is essential for proliferation of adenomatous polyposis coli (APC)-deficient cells. Rapamycin targets eEF2 indirectly through the mTORC1-S6K-eEF2K pathway, and treatment of APC-deficient adenomas with rapamycin induces tumor cell growth arrest and differentiation [27].
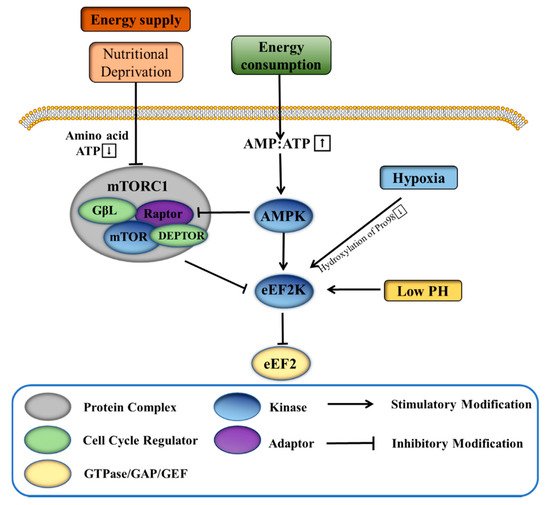
The rapid proliferation of tumor cells also requires a large amount of oxygen, which frequently is overdrawn enough to induce a hypoxic environment. It has been found that hypoxia inhibits protein synthesis in breast cancer cells in part through 4E-BP1 and the eEF2K pathway, controlled by mTOR [28]. Additionally, eEF2K is activated and induces eEF2 phosphorylation during hypoxia independent of AMPK and mTORC1 signaling [29]. The eEF2K residue Pro98 is a generally conserved linker between the calmodulin binding domain and the catalytic domain, and when it is hydroxylated, the binding of calmodulin to eEF2K is reduced and the activity of eEF2K is significantly limited. Under normoxia, proline hydroxylase catalyzes the hydroxylation of Pro98 of eEF2K, thus inhibiting eEF2K activity. However, when the cells are hypoxic, the activity of proline hydroxylase is inhibited, thereby releasing the normal inhibition of eEF2K (Figure 1) [29]. Normal cells rely on mitochondrial oxidative phosphorylation to produce energy, while tumor cells mainly generate energy through glycolysis under hypoxic/normoxic conditions (Warburg effect) [30]. The upregulation of eEF2K accelerates glycolysis to promote human breast cancer cells in development and progression. eEF2K inhibits protein phosphatase 2A-A (PP2A-A) synthesis, thereby interfering with its promotion of c-Myc ubiquitin-proteasome degradation, and finally activates the transcription of pyruvate kinase M2 subtype (PKM2) to promote glycolysis [31].
Low pH is frequently a major feature of tumor microenvironments. In tumor cells, unrestricted glycolysis leads to a large accumulation of lactic acid, thereby acidifying the local environment [32][33]. Under acidic conditions, or low pH, overexpression of eEF2K inhibits protein synthesis. At neutral pH, by contrast, overexpression of eEF2K does not affect protein synthesis, indicating the activation of eEF2K in acidic conditions (Figure 1) [34]. However, the activation of eEF2K is independent of the activity of mTORC1. It is understood that the affinity of eEF2K to CaM is enhanced at acidic pH, and the histidine residue (H108) in CaM is essential for the activation of eEF2K [35].
Overall, the activity of eEF2K is known to change with the conditions of the tumor microenvironment (e.g., energy deficiency, hypoxia, low pH), thereby regulating the process of tumor protein synthesis and ultimately protecting the survival of tumor cells in otherwise harsh conditions.
2.2. eEF2K Inhibits Cell Apoptosis
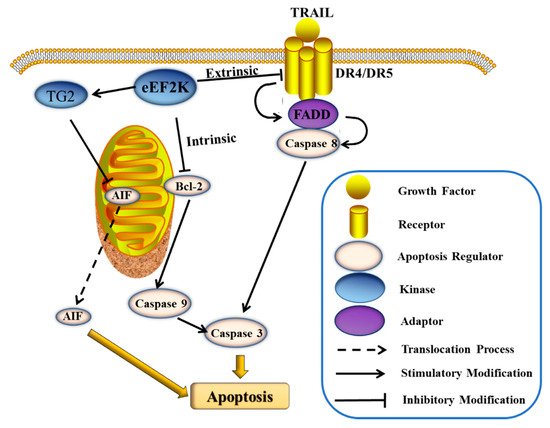
Cell apoptosis is typically dysregulated in cancers, which leads to rampant proliferation. Accordingly, inducing apoptosis in cancer cells is an important mechanism of anti-tumor drugs. The expression of eEF2K can inhibit apoptosis and promote cancer cell survival [4]. The suppression of this mechanism would enable eEF2K to be used mechanistically as a new drug target for single or multi-agent cancer chemotherapeutics. The caspases represent an important family of proteins that regulate cell apoptosis. Among them, caspase 8 and caspase 9 are the key regulatory proteins in extrinsic and intrinsic apoptotic pathways, respectively [36]. The cleavage of these caspases will eventually lead to the cleavage of caspase 3 and ultimately apoptosis of the cell [37]. Some eEF2K inhibitors have been shown to induce tumor cell apoptosis by these mediated extrinsic and/or intrinsic apoptosis pathways. The cleavage of caspase 8 that is induced by tumor necrosis factor (TNF) family proteins is an important aspect of the extrinsic apoptosis pathway [38]. The TNF-related apoptosis-inducing ligand (TRAIL) belongs to the TNF family. TRAIL can bind to the death receptors DR4 and DR5 to form the death-inducing signaling complex (DISC) and to upregulate Fas-associated protein with death domain (FADD), thereby inducing caspase-8-dependent apoptosis (Figure 2) [39]. Treatment of glioma cells with the eEF2K inhibitor, NH125 (1), showed the enhancement of TRAIL-induced apoptosis, and, with the increase of dosed NH125, the cleaved PARP and caspase 8 levels increased significantly [40]. Bcl-2 is another important family of proteins that regulate endogenous apoptosis [41]. The founding member protein Bcl-2 and Bcl-xL are anti-apoptotic proteins in the Bcl-2 family, and NH125 down-regulates the expression of Bcl-xL in glioma cells [40]. It was additionally shown that silencing eEF2K induces caspase-9 cleavage and Bcl-2 downregulation in breast cancer cells (Figure 2) [42]. Meanwhile, inhibiting eEF2K enhances the effect of doxorubicin in an orthotopic model of breast cancer [42]. Furthermore, eEF2K is highly expressed in pancreatic cancer (PaCa) and acts to inhibit apoptosis [14]. Treatment of PaCa cells with the natural product inhibitor of eEF2K, rottlerin (29), not only induces the collapse of mitochondrial potential causing intrinsic apoptosis, but also causes extrinsic apoptosis regulated by TRAIL and caspase 8 [14]. At the same time, rottlerin also effects the expression of TG2 (PKC-δ/tissue transglutaminase), which in turn activates apoptosis-inducing factor (AIF), and ultimately causes caspase-dependent apoptosis (Figure 2) [14].
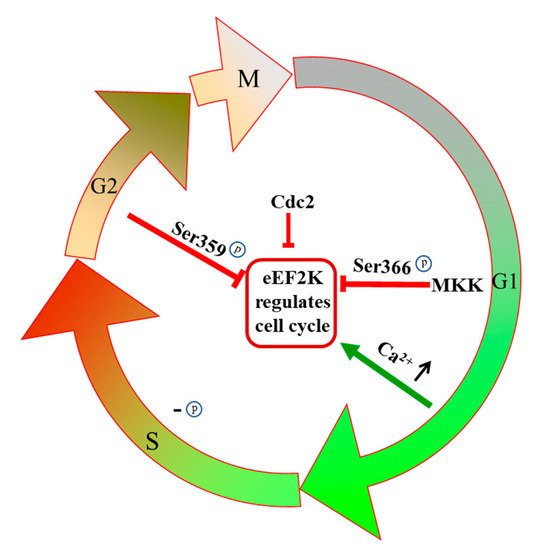
2.3. eEF2K Regulates the Cell Cycle
The cell cycle is inextricably tied to protein synthesis, and thus the impact of eEF2K in elongation can be understandably expanded. For one thing, phosphorylation of eEF2K residues Ser359 and Ser366 leads to its inactivation, which accordingly regulates cell cycle progression [26]. The inhibition of eEF2K has been shown to arrest breast cancer cells at the G0/G1-S phase [43]. Conversely, eEF2K is inactivated during G1 cell growth, so eEF2 becomes active and promotes protein synthesis [43]. The Ser366 residue of eEF2K is a point of regulation by MKK and mTOR pathways, and when cells enter the G1 phase, Ser366 is phosphorylated rapidly in an MKK-dependent manner (Figure 3) [44]. During S phase DNA replication, eEF2K is slowly dephosphorylated, and eEF2 activity is eventually inhibited completely during G2 and mitosis (Figure 3) [43]. It is well-known that eEF2K is a calcium/calmodulin-dependent protein kinase, and calcium binding to CaM has an important influence on the process of mitosis [5][45]. In the G1/S transition, the intracellular Ca2+ concentration increases. Under environmental conditions of high calcium concentration, calmodulin can bind to eEF2K and activate it so that the cells enter the S phase (Figure 3) [46]. Another function of cellular calcium is to upregulate cAMP levels [47]. The cAMP in turn activates PKA, which can activate eEF2K by phosphorylation of the Ser500 residue [47][48]. The regulation of eEF2K on the G2/M phase is related to the phosphorylation at the Ser359 residue, and this leads to inhibition of eEF2K activity independent of Ca2+ concentration (Figure 3) [49]. It has been shown that eEF2K can also be regulated by cell cycle-related proteins to affect cell cycle progression. For example, human cyclin-dependent kinase 1 (CDC2) is regulated by mTORC1 and gets activated in the early stage of mitosis; it then inactivates eEF2K, and thus protein synthesis is carried out in mitotic cells (Figure 3) [50]. Cell cycle progression is closely related to proliferation in tumor cells, and chemical interference can accordingly inhibit proliferation and eventually lead to cell death [51]. The successful launch of the CDK4 and CDK6 targeting cell cycle inhibitor palbociclib, which was approved by the U.S. FDA in 2015, has encouraged further research on cell cycle-regulating anticancer agents for use as drugs [52]. Since eEF2K has a regulatory effect on multiple links in the cell cycle process, this represents an attractive new target for future cancer treatments.
2.4. eEF2K Regulates Cell Autophagy
Autophagy is a “self-consuming” program of cells that is used to remove damage and dysfunction or unnecessary proteins, and it is closely associated with human diseases such as cancer [53][54]. Under conditions of starvation or stress, excess cells may undergo autophagy so that the remaining cells can better survive [55][56]. Autophagy mechanistically works by lysosomes in cells degrading their own organelles and other macromolecules, and it is an important process for eukaryotes to carry out the turnover of intracellular substances [57][58]. As previously discussed, tumor cell proliferation often results in microenvironment nutritional starvation or stress conditions. Autophagy can protect cells from apoptosis and promotes tumor progression [57][59]. mTOR is an important regulator of autophagy and the upstream protein of eEF2K. Previous studies have demonstrated that eEF2K induces autophagy to protect cancer cells survival [60][61]. For example, it was found that during amino acid starvation and endoplasmic reticulum (ER) stress, eEF2K is activated to induce autophagy [62][63]. Inhibiting eEF2K-mediated autophagy has been reported to enhance the antitumor effect of MK-2206, an AKT inhibitor, on human nasopharyngeal carcinoma and human glioma cells [60][61]. The induction of autophagy is mediated via the TSC2/mTOR/S6 kinase/eEF-2 kinase pathway [60]. It was also found that silencing of eEF2K can inhibit autophagy through the mTORC1/p70S6K signaling pathway and increase the sensitivity of human glioma cells to 2-deoxy-d-glucose (2-DG) [64].
However, in contrast to the above outcome in human nasopharyngeal carcinoma and human glioma cells, it was found that silencing of eEF2K activity can induce autophagy to promote the proliferation of colon cancer cells but not enhance the anticancer effect of MK-2206 in human colon cancer cells [65]. The negative regulation of eEF2K on autophagy in colon cancer cells is dependent on the activation of the AMPK-ULK1 pathway [65]. Another experiment on human lung cancer cells showed that eEF2K protects cell survival under nutrient deprivation, but this effect is due to its inhibition of protein synthesis rather than regulation of autophagy [15]. Thus, the particular cancer cell type and specific mechanism of action being observed is important to consider.
2.5. eEF2K Promotes Tumor Angiogenesis, Metastasis, and Invasion
Angiogenesis, the growth of new blood vessels, provides tumors with more nutrients to promote their growth, and plays a key role in tumor proliferation, metastasis, and invasion [66][67][68]. Overexpression of eEF2K has been shown to promote angiogenesis, invasion, and metastasis in multiples types of tumors [69][70]. Inhibition of eEF2K expression can likewise prevent these tumor processes. For example, knockdown of eEF2K was found to prevent tumor progression and angiogenesis of hepatocellular carcinoma via the PI3K/Akt and STAT3 signaling pathway [11]. In triple-negative breast cancer (TNBC) cells, the proto-oncogene transcription factor forkhead box M1 (FOXM1) can regulate eEF2K and affect breast cancer cell migration and invasion, progression, and tumorigenesis [18]. The dual inhibitory effect of microRNA-34a on the FOXM1/eEF2K axis can regulate the growth and invasion of TNBC [71]. In addition, TNBC with mutations in PTEN and p53 is more sensitive to eEF2K inhibitors, and this effect is related to the AKT signaling pathway [8]. Similarly, it was found after knocking out eEF2K that the invasion and metastasis of lung cancer cells was inhibited [21]. Proud et al. found that this inhibitory effect may be related to integrin signaling proteins to control cell–cell/cell–extracellular matrix interactions and cell mobility [72].
References
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Clin. Pharmacol. 2018, 834, 188–196.
- Rosenzweig, S.A. Chapter Three—Acquired Resistance to Drugs Targeting Tyrosine Kinases. Adv. Cancer. Res. 2018, 138, 71–98.
- Cote, G.P.; Luo, X.; Murphy, M.B.; Egelhoff, T.T. Mapping of the novel protein kinase catalytic domain of dictyostelium myosin II heavy chain kinase A. J. Biol. Chem. 1997, 272, 6846–6849.
- Kenney, J.W.; Moore, C.E.; Wang, X.; Proud, C.G. Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv. Biol. Regul. 2014, 55, 15–27.
- Proud, C.G. Regulation and roles of elongation factor 2 kinase. Biochem. Soc. Trans. 2015, 43, 328–332.
- Yamaguchi, H.; Matsushita, M.; Nairn, A.C.; Kuriyan, J. Crystal structure of the atypical protein kinase domain of a TRP channel with phosphotransferase activity. Mol. Cell 2001, 7, 1047–1057.
- Delaidelli, A.; Khan, D.; Leprivier, G.; Pfister, S.M.; Taylor, M.D.; Maris, J.M.; Sorensen, P. OS5-173 Inhibition of eEF2K as a novel therapeutic strategy in neuroblastoma and medulloblastoma. Can. J. Neurol. Sci. 2016, 43, S3.
- Liu, J.C.; Voisin, V.; Wang, S.; Wang, D.Y.; Jones, R.A.; Datti, A.; Uehling, D.; Al-Awar, R.; Egan, S.E.; Bader, G.D. Combined deletion of Pten and p53 in mammary epithelium accelerates triple-negative breast cancer with dependency on eEF2K. EMBO Mol. Med. 2015, 6, 1542–1560.
- Gassart, A.D.; Martinon, F. Translating the anticancer properties of eEF2K. Cell Cycle 2017, 16, 299–300.
- Zhang, H.; Bialkowska, A.; Rusovici, R.; Chanchevalap, S.; Shim, H.; Katz, J.P.; Yang, V.W.; Yun, C.C. Lysophosphatidic acid facilitates proliferation of colon cancer cells via induction of Krüppel-like factor 5. J. Biol. Chem. 2007, 282, 15541–15549.
- Zhou, Y.; Li, Y.; Xu, S.; Lu, J.; Zhu, Z.; Chen, S.; Tan, Y.; He, P.; Xu, J.; Proud, C.G.; et al. Eukaryotic elongation factor 2 kinase promotes angiogenesis in hepatocellular carcinoma via PI3K/Akt and STAT3. Int. J. Cancer. 2020, 146, 1383–1395.
- Horman, S.; Beauloye, C.; Vertommen, D.; Vanoverschelde, J.L.; Hue, L.; Rider, M.H. Myocardial ischemia and increased heart work modulate the phosphorylation state of eukaryotic elongation factor-2. J. Biol. Chem. 2003, 278, 41970–41976.
- Karakas, D.; Ozpolat, B. Eukaryotic elongation factor-2 kinase (eEF2K) signaling in tumor and microenvironment as a novel molecular target. J. Mol. Med. 2020, 98, 775–787.
- Ashour, A.A.; Abdel-Aziz, A.A.H.; Mansour, A.M.; Alpay, S.N.; Huo, L.F.; Ozpolat, B. Targeting elongation factor-2 kinase (eEF-2K) induces apoptosis in human pancreatic cancer cells. Apoptosis 2014, 19, 241–258.
- Moore, C.E.; Wang, X.; Xie, J.; Pickford, J.; Barron, J.; Regufe da Mota, S.; Versele, M.; Proud, C.G. Elongation factor 2 kinase promotes cell survival by inhibiting protein synthesis without inducing autophagy. Cell Signal. 2016, 28, 284–293.
- Leprivier, G.; Rotblat, B.; Khan, D.; Jan, E.; Sorensen, P.H. Stress-mediated translational control in cancer cells. Biochim. Biophys. Acta 2015, 1849, 845–860.
- Russnes, H.G.; Caldas, C. eEF2K-a new target in breast cancers with combined inactivation of p53 and PTEN. EMBO Mol. Med. 2014, 6, 1512–1514.
- Hamurcu, Z.; Ashour, A.; Kahraman, N.; Ozpolat, B. FOXM1 regulates expression of eukaryotic elongation factor 2 kinase and promotes proliferation, invasion and tumorgenesis of human triple negative breast cancer cells. Oncotarget 2016, 7, 16619–16635.
- Shi, N.; Chen, X.; Liu, R.; Wang, D.; Su, M.; Wang, Q.; He, A.; Gu, H. Eukaryotic elongation factors 2 promotes tumor cell proliferation and correlates with poor prognosis in ovarian cancer. Tissue Cell 2018, 53, 53–60.
- Bayraktar, R.; Pichler, M.; Kanlikilicer, P.; Ivan, C.; Bayraktar, E.; Kahraman, N.; Aslan, B.; Oguztuzun, S.; Ulasli, M.; Arslan, A.; et al. MicroRNA 603 acts as a tumor suppressor and inhibits triple-negative breast cancer tumorigenesis by targeting elongation factor 2 kinase. Oncotarget 2017, 8, 11641–11658.
- Bircan, H.A.; Gurbuz, N.; Pataer, A.; Caner, A.; Kahraman, N.; Bayraktar, E.; Bayraktar, R.; Erdogan, M.A.; Kabil, N.; Ozpolat, B. Elongation factor-2 kinase (eEF-2K) expression is associated with poor patient survival and promotes proliferation, invasion and tumor growth of lung cancer. Lung Cancer 2018, 124, 31–39.
- Leprivier, G.; Remke, M.; Rotblat, B.; Dubuc, A.; Mateo, A.R.; Kool, M.; Agnihotri, S.; El-Naggar, A.; Yu, B.; Somasekharan, S.P.; et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 2013, 153, 1064–1079.
- Jewell, J.L.; Guan, K.L. Nutrient signaling to mTOR and cell growth. Trends Biochem. Sci. 2013, 38, 233–242.
- Ryazanov, A.G.; Shestakova, E.A.; Natapov, P.G. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature 1988, 334, 170–173.
- Proud, C.G. mTORC1 regulates the efficiency and cellular capacity for protein synthesis. Biochem. Soc. Trans. 2013, 41, 923–926.
- Proud, C.G. Signalling to translation: How signal transduction pathways control the protein synthetic machinery. Biochem. J. 2007, 403, 217–234.
- Faller, W.J.; Jackson, T.J.; Knight, J.R.; Ridgway, R.A.; Jamieson, T.; Karim, S.A.; Jones, C.; Radulescu, S.; Huels, D.J.; Myant, K.B.; et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 2015, 517, 497–500.
- Connolly, E.; Braunstein, S.; Formenti, S.; Schneider, R.J. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol. Cell. Biol. 2006, 26, 3955–3965.
- Moore, C.E.J.; Mikolajek, H.; Sergio, R.D.M.; Wang, X.; Kenney, J.W.; Werner, J.R.M.; Proud, C.G.J.M.; Biology, C. Elongation Factor 2 Kinase Is Regulated by Proline Hydroxylation and Protects Cells during Hypoxia. Mol. Cell. Biol. 2015, 35, 1788–1804.
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033.
- Cheng, Y.; Ren, X.; Yuan, Y.; Shan, Y.; Li, L.; Chen, X.; Zhang, L.; Takahashi, Y.; Yang, J.W.; Han, B.; et al. eEF-2 kinase is a critical regulator of Warburg effect through controlling PP2A-A synthesis. Oncogene 2016, 35, 6293–6308.
- Helmlinger, G.; Schell, A.; Dellian, M.; Forbes, N.S.; Jain, R.K. Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin. Cancer Res. 2002, 8, 1284–1291.
- Shime, H.; Yabu, M.; Akazawa, T.; Kodama, K.; Matsumoto, M.; Seya, T.; Inoue, N. Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J. Immunol. 2008, 180, 7175–7183.
- Dorovkov, M.V.; Pavur, K.S.; Petrov, A.G. Regulation of elongation factor-2 kinase by pH. Biochemistry 2002, 41, 13444–13450.
- Xie, J.; Mikolajek, H.; Pigott, C.R.; Hooper, K.J.; Mellows, T.; Moore, C.E.; Mohammed, H.; Werner, J.M.; Thomas, G.J.; Proud, C.G. Molecular mechanism for the control of eukaryotic elongation factor 2 kinase by pH: Role in cancer cell survival. Mol. Cell. Biol. 2015, 35, 1805–1824.
- Theodoropoulos, G.E.; Gazouli, M.; Vaiopoulou, A.; Leandrou, M.; Nikouli, S.; Vassou, E.; Kouraklis, G.; Nikiteas, N. Polymorphisms of Caspase 8 and Caspase 9 gene and colorectal cancer susceptibility and prognosis. Int. J. Colorectal. Dis. 2011, 26, 1113–1118.
- Park, H.S.; Jun, D.Y.; Han, C.R.; Kim, Y.H.J.B.B.R.C. Protein tyrosine kinase p56lck-deficiency confers hypersusceptibility to rho-fluorophenylalanine (pFPhe)-induced apoptosis by augmenting mitochondrial apoptotic pathway in human Jurkat T cells. Biochem. Biophys. Res. Commun. 2008, 377, 280–285.
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703.
- Bellail, A.C.; Tse, M.C.L.; Song, J.H.; Phuphanich, S.; Olson, J.J.; Sun, S.Y.; Hao, C.H. DR5-mediated DISC controls caspase-8 cleavage and initiation of apoptosis in human glioblastomas. J. Cell. Mol. Med. 2010, 14, 1303–1317.
- Zhang, Y.; Cheng, Y.; Zhang, L.; Ren, X.C.; Huber-Keener, K.J.; Lee, S.; Yun, J.; Wang, H.G.; Yang, J.M. Inhibition of eEF-2 kinase sensitizes human glioma cells to TRAIL and down-regulates Bcl-xL expression. Biochem. Biophys. Res. Commun. 2011, 414, 129–134.
- Van Loo, G.; Saelens, X.; van Gurp, M.; MacFarlane, M.; Martin, S.J.; Vandenabeele, P. The role of mitochondrial factors in apoptosis: A Russian roulette with more than one bullet. Cell Death Differ. 2002, 9, 1031–1042.
- Tekedereli, I.; Alpay, S.N.; Tavares, C.D.J.; Cobanoglu, Z.E.; Kaoud, T.S.; Sahin, I.; Sood, A.K.; Lopez-Berestein, G.; Dalby, K.N.; Ozpolat, B. Targeted Silencing of Elongation Factor 2 Kinase Suppresses Growth and Sensitizes Tumors to Doxorubicin in an Orthotopic Model of Breast Cancer. PLoS ONE 2012, 7, e41171.
- Parmer, T.G.; Ward, M.D.; Yurkow, E.J.; Vyas, V.H.; Kearney, T.J.; Hait, W.N. Activity and regulation by growth factors of calmodulin-dependent protein kinase III (elongation factor 2-kinase) in human breast cancer. Br. J. Cancer 1999, 79, 59–64.
- Roberts, E.C.; Hammond, K.; Traish, A.M.; Resing, K.A.; Ahn, N.G. Identification of G2/M targets for the MAP kinase pathway by functional proteomics. Proteomics 2010, 6, 4541–4553.
- Ratan, R.R.; Maxfield, F.R.; Shelanski, M.L. Long-lasting and rapid calcium changes during mitosis. J. Cell Biol. 1988, 107, 993–999.
- Santella, L. The role of calcium in the cell cycle: Facts and hypotheses. Biochem. Biophys. Res. Commun. 1998, 244, 317–324.
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21.
- Gutzkow, K.B.; Låhne, H.U.; Naderi, S.; Torgersen, K.M.; Skålhegg, B.; Koketsu, M.; Uehara, Y.; Blomhoff, H.K. Cyclic AMP inhibits translation of cyclin D3 in T lymphocytes at the level of elongation by inducing eEF2-phosphorylation. Cell. Signal. 2003, 15, 871–881.
- Pyr Dit Ruys, S.; Wang, X.; Smith, E.M.; Herinckx, G.; Hussain, N.; Rider, M.H.; Vertommen, D.; Proud, C.G. Identification of autophosphorylation sites in eukaryotic elongation factor-2 kinase. Biochem. J. 2012, 442, 681–692.
- Smith, E.M.; Proud, C.G. cdc2–cyclin B regulates eEF2 kinase activity in a cell cycle- and amino acid-dependent manner. EMBO J. 2008, 27, 1005–1016.
- Lee, B.; Sandhu, S.; Mcarthur, G. Cell cycle control as a promising target in melanoma. Curr. Opin. Oncol. 2015, 27, 141–150.
- Petroni, G.; Formenti, S.C.; Chen-Kiang, S.; Galluzzi, L. Immunomodulation by anticancer cell cycle inhibitors. Nat. Rev. Immunol. 2020, 20, 669–679.
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42.
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075.
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734.
- Cheng, Y.; Li, H.; Ren, X.; Niu, T.; Hait, W.N.; Yang, J. Cytoprotective Effect of the Elongation Factor-2 Kinase-Mediated Autophagy in Breast Cancer Cells Subjected to Growth Factor Inhibition. PLoS ONE 2010, 5, e9715.
- Jung, S.; Jeong, H.; Yu, S.W. Autophagy as a decisive process for cell death. Exp. Mol. Med. 2020, 52, 921–930.
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046.
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410.
- Cheng, Y.; Ren, X.; Zhang, Y.; Patel, R.; Sharma, A.; Wu, H.; Robertson, G.P.; Yan, L.; Rubin, E.; Yang, J.M. eEF-2 kinase dictates cross-talk between autophagy and apoptosis induced by Akt Inhibition, thereby modulating cytotoxicity of novel Akt inhibitor MK-2206. Cancer Res. 2011, 71, 2654–2663.
- Zhao, Y.Y.; Tian, Y.; Liu, L.; Zhan, J.H.; Hou, X.; Chen, X.; Zhou, T.; Huang, Y.; Zhang, L. Inhibiting eEF-2 kinase-mediated autophagy enhanced the cytocidal effect of AKT inhibitor on human nasopharyngeal carcinoma. Drug Des. Dev. Ther. 2018, 12, 2655–2663.
- Py, B.F.; Boyce, M.; Yuan, J. A critical role of eEF-2K in mediating autophagy in response to multiple cellular stresses. Autophagy 2009, 5, 393–396.
- Boyce, M.; Py, B.F.; Ryazanov, A.G.; Minden, J.S.; Long, K.; Ma, D.; Yuan, J. A pharmacoproteomic approach implicates eukaryotic elongation factor 2 kinase in ER stress-induced cell death. Cell Death Differ. 2008, 15, 589–599.
- Wu, H.; Zhu, H.; Liu, D.X.; Niu, T.K.; Ren, X.; Patel, R.; Hait, W.N.; Yang, J.M. Silencing of elongation factor-2 kinase potentiates the effect of 2-deoxy-D-glucose against human glioma cells through blunting of autophagy. Cancer Res. 2009, 69, 2453–2460.
- Xie, C.M.; Liu, X.Y.; Sham, K.W.; Lai, J.M.; Cheng, C.H. Silencing of EEF2K (eukaryotic elongation factor-2 kinase) reveals AMPK-ULK1-dependent autophagy in colon cancer cells. Autophagy 2014, 10, 1495–1508.
- Chen, C.; Xu, Z.Q.; Zong, Y.P.; Ou, B.C.; Shen, X.H.; Feng, H.; Zheng, M.H.; Zhao, J.K.; Lu, A.G. CXCL5 induces tumor angiogenesis via enhancing the expression of FOXD1 mediated by the AKT/NF-κB pathway in colorectal cancer. Cell Death Dis. 2019, 10, 178.
- Hoshi, T.; Watanabe Miyano, S.; Watanabe, H.; Sonobe, R.M.K.; Seki, Y.; Ohta, E.; Nomoto, K.; Matsui, J.; Funahashi, Y. Lenvatinib induces death of human hepatocellular carcinoma cells harboring an activated FGF signaling pathway through inhibition of FGFR–MAPK cascades. Biochem. Biophys. Res. Commun. 2019, 513, 1–7.
- Zhu, H.; Song, H.; Chen, G.; Yang, X.; Liu, J.; Ge, Y.; Lu, J.; Qin, Q.; Zhang, C.; Xu, L.; et al. eEF2K promotes progression and radioresistance of esophageal squamous cell carcinoma. Radiother. Oncol. 2017, 124, 439–447.
- Shi, Q.; Xu, X.; Liu, Q.; Luo, F.; Shi, J.; He, X. MicroRNA-877 acts as a tumor suppressor by directly targeting eEF2K in renal cell carcinoma. Oncol. Lett. 2016, 11, 1474–1480.
- Ashour, A.A.; Gurbuz, N.; Alpay, S.N.; Abdel-Aziz, A.A.H.; Mansour, A.M.; Huo, L.; Ozpolat, B. Elongation factor-2 kinase regulates TG2/β1 integrin/Src/uPAR pathway and epithelial-mesenchymal transition mediating pancreatic cancer cells invasion. J. Cell. Mol. Med. 2014, 18, 2235–2251.
- Bayraktar, R.; Ivan, C.; Bayraktar, E.; Kanlikilicer, P.; Kabil, N.N.; Kahraman, N.; Mokhlis, H.A.; Karakas, D.; Rodriguez-Aguayo, C.; Arslan, A.; et al. Dual Suppressive Effect of miR-34a on the FOXM1/eEF2-Kinase Axis Regulates Triple-Negative Breast Cancer Growth and Invasion. Clin. Cancer Res. 2018, 24, 4225–4241.
- Xie, J.; Shen, K.; Lenchine, R.V.; Gethings, L.A.; Trim, P.J.; Snel, M.F.; Zhou, Y.; Kenney, J.W.; Kamei, M.; Kochetkova, M.; et al. Eukaryotic elongation factor 2 kinase upregulates the expression of proteins implicated in cell migration and cancer cell metastasis. Int. J. Cancer 2018, 142, 1865–1877.

