 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrew Eisen | + 1039 word(s) | 1039 | 2021-04-21 05:22:01 | | | |
| 2 | Catherine Yang | Meta information modification | 1039 | 2021-04-22 03:33:39 | | | | |
| 3 | Conner Chen | Meta information modification | 1039 | 2021-10-12 05:19:34 | | |
Video Upload Options
The site of origin of amyotrophic lateral sclerosis (ALS), although unsettled, is increasingly recognized as being cortico-fugal, which is a dying-forward process primarily starting in the corticomotoneuronal system.
1. Introduction
Neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS), are complex polygenic diseases, resulting in multisystem impairment of neocortical networks, primarily involving the neocortex. Specific to ALS is dysfunction of the expanded human corticomotoneuronal system [1]. This system is the anatomical infrastructure of many early clinical features of ALS, a singularly human disorder [2][3]. Early deficits include loss of vocalization requiring the integration of a complex respiratory system, impaired fractionation of digits and thumb opposability, responsible for manipulative agility, and difficulty with upright walking, especially the ability to navigate uneven and tricky surfaces. These initial symptoms reflect dysfunction of the corticomotoneuronal system [4]. In addition is the association of frontotemporal dementia (FTD), causing language impairment, failing executive function, and deteriorating socialization [5][6]. This paragraph underscores my present view that ALS is a “brain disease” [3].
In coining the term “corticomotoneuronal hypothesis” [7], I first proposed some thirty years ago that ALS had its origins in the brain. This resulted in considerable controversy and many ALS physicians and scientists regarded the idea with skepticism. Though the earliest anatomical site of neurodegeneration in ALS is still not known for certain, as we enter the third decade of the 21st century, a top-down, dying-forward process, as opposed to a bottom-up, dying-back process, has become increasingly adopted. The dying-forward hypothesis proposes that glutamate excitotoxicity at the level of the cortical motor neuron ultimately results in anterior horn cell metabolic deficit. In contrast, the dying-back hypothesis proposes that ALS pathology is initiated at the level of the lower motor neurons and advances in a retrograde direction from the neuromuscular junction into the central nervous system. See Figure 1 taken from Kiernan et al. 2011 [2].
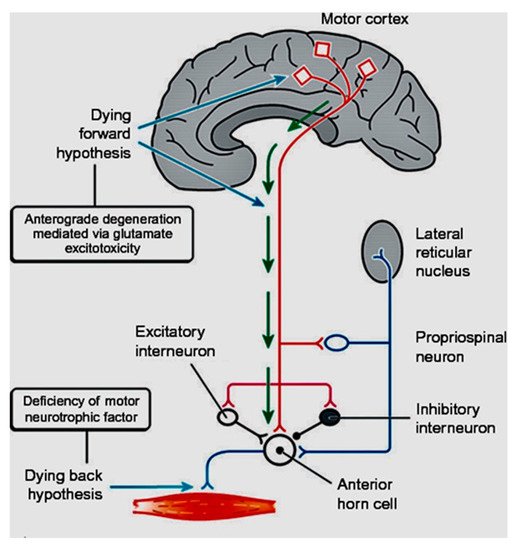
Figure 1. From Kiernan et al. [2]. The dying-forward hypothesis postulates that ALS commences in the motor and pre-motor cortices’ pyramidal neurons and, through antegrade mechanisms, causes dysfunction and death of the bulbar and spinal motor neurons. Excitotoxicity is important but not the only factor. The hallmark TAR DNA-binding protein 43 (TDP-43) pathology, seen in >95% of patients with ALS, is largely restricted to corticofugal projecting neurons (“dying forward”). In broader terms, this site of origin may be considered as the nidus of a spreading network disorder associated with frontotemporal dementia in ALS. In any event, ALS is best regarded as a degenerative brain disease. The figure indicates that there are alternative hypotheses of origin site which include dying-back and independent degeneration of the upper and lower motor neurons.
2. The Growth of Electrophysiological Support
Prior to the latter half of the 1980s, investigative work in living ALS patients throughout much of the world was biased towards the lower motor neuron. Function and dysfunction of the ALS lower motor neuron was readily accessible, primarily using electromyography [8][9]. This lower motor neuron-centric view of ALS further emphasized the disorder being erroneously classified as a neuromuscular disease. Ability to examine the upper motor neuron in vivo before the 1990s was limited, and clinical assessment of upper motor neuron deficits in ALS continues to be fraught with difficulties [10][11]. Now that it is unanimously accepted that ALS is a multisystem disorder, with prominent cortical involvement, a view fully supported by overwhelming evidence from psychometric, imaging, genetic, and pathological studies, it is untenable in the twenty-first century to continue to classify ALS as a neuromuscular disorder [12]. It should correctly be classified as a neurodegenerative disease, as was suggested two decades ago by Eisen and Calne [13].
My early formulations proposing the primacy of the brain in ALS developed from two thoughts [7][13]. First, as espoused by Hudson and Kiernan [14], related to the selective sparing of certain motor neuron pools in ALS. In particular, those anterior horn cells that do not receive direct (monosynaptic) input from the upper motor neuron (corticomotoneurons), a term initially coined by Bernhard et al. (1954). The sparing of these motor neuron pools is relative, as eventually they too are affected. However, various types of oculomotor dysfunction such as square-wave jerks, saccadic dysmetria, abnormal cogwheeling smooth pursuit, and head shaking, and positional nystagmus have been described in ALS patients at a relatively early disease stage [15]. But, since it is generally established that oculomotor motoneurons do not receive direct projections from the cortex, if ALS results from pathological processes involving such direct projections, then it is important to determine whether there is oculomotor weakness in patients with ALS. Clinical and neurophysiological findings demonstrate that while there are deficits, as those described above, these reflect problems with higher-level control and are not indicative of the motoneuron loss or weakness that characterises ALS in, for example, the hand and the foot [16].
Similarly, electrophysiological studies have shown that the external anal sphincter is not normal in ALS, but there is relative resistance sufficient to prevent incontinence, even in the longer-surviving older patients [17]. My other thought regarding the primacy of the brain in ALS arose from the fact that neurodegenerative disorders, certainly ALS, are uniquely human. There are no natural animal models that come close to human ALS. I accept the considerable value that induced animal models have and continue to play in furthering our understanding of ALS, but they cannot mimic the human disease. This, I contended, is largely related to the expanded human neocortex. In particular, the direct corticospinal projection (corticomotoneuronal system) is the latest in the development of the nervous system being associated with the least well conserved genes and therefore, most vulnerable to environmental change.
In ALS, it only became feasible to study the upper motor neuron in awake humans, in vivo, late in the 1980s. During this period, MRI studies of ALS were sparse, limited to very few patients, and the results were non-specific [18][19]. After 2000, enhanced software programs began to give insight into several aspects of ALS [20]. In the late 1980s, transcranial magnetic stimulation (TMS) started to be used to investigate the upper motor neuron in ALS, first in the United Kingdom [21] and subsequently in Canada [22][23][24]. Upper motor neuron ALS physiological studies using TMS subsequently employed increasingly sophisticated techniques: triple stimulation, peristimulus time histograms, and finally, threshold tracking developed in Australia [25][26][27][28]. This last technique has consistently shown that hyperexcitability precedes the onset of clinically overt ALS [29].
References
- Lemon, R.N.; Griffiths, J. Comparing the function of the corticospinal system in different species: Organizational differences for motor specialization? Muscle Nerve 2005, 32, 261–279.
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955.
- Eisen, A. Amyotrophic lateral sclerosis: A 40-year personal perspective. J. Clin. Neurosci. 2009, 16, 505–512.
- Lemon, R.N. Descending pathways in motor control. Annu. Rev. Neurosci. 2008, 31, 195–218.
- Snowden, J.S.; Harris, J.; Richardson, A.; Rollinson, S.; Thompson, J.C.; Neary, D.; Mann, D.M.; Pickering-Brown, S. Frontotemporal dementia with amyotrophic lateral sclerosis: A clinical comparison of patients with and without repeat expansions in C9orf72. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 172–176.
- Woolley, S.C.; Strong, M.J. Frontotemporal Dysfunction and Dementia in Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 787–805.
- Eisen, A.; Kim, S.; Pant, B. Amyotrophic lateral sclerosis (ALS): A phylogenetic disease of the corticomotoneuron? Muscle Nerve 1992, 15, 219–224.
- Marinacci, A.A.; VonHagen, K.O. Electromyography in amyotrophic lateral sclerosis. A review. Bull. Los Angel. Neurol. Soc. 1974, 39, 17–29.
- Daube, J.R. Electrophysiologic studies in the diagnosis and prognosis of motor neuron diseases. Neurol. Clin. 1985, 3, 473–493.
- Swash, M. Why are upper motor neuron signs difficult to elicit in amyotrophic lateral sclerosis? J. Neurol. Neurosurg. Psychiatry 2012, 83, 659–662.
- Swash, M.; Burke, D.; Turner, M.R.; Grosskreutz, J.; Leigh, P.N.; deCarvalho, M.; Kiernan, M.C. Occasional essay: Upper motor neuron syndrome in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2020, 91, 227–234.
- van Es, M.A.; Goedee, H.S.; Westeneng, H.J.; Nijboer, T.C.W.; van den Berg, L.H. Is it accurate to classify ALS as a neuromuscular disorder? Expert Rev. Neurother. 2020, 20, 895–906.
- Eisen, A.; Calne, D. Amyotrophic lateral sclerosis, Parkinson’s disease and Alzheimer’s disease: Phylogenetic disorders of the human neocortex sharing many characteristics. Can. J. Neurol. Sci. 1992, 19, 117–123.
- Hudson, A.J.; Kiernan, J.N. Preservation of certain voluntary muscles in motoneurone disease. Lancet 1988, 1, 652–653.
- Kang, B.H.; Kim, J.I.; Lim, Y.M.; Kim, K.K. Abnormal Oculomotor Functions in Amyotrophic Lateral Sclerosis. J. Clin. Neurol. 2018, 14, 464–471.
- Eisen, A.; Braak, H.; Del Tredici, K.; Lemon, R.; Ludolph, A.C.; Kiernan, M.C. Cortical influences drive amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 917–924.
- Carvalho, M.; Schwartz, M.S.; Swash, M. Involvement of the external anal sphincter in amyotrophic lateral sclerosis. Muscle Nerve 1995, 18, 848–853.
- Goodin, D.S.; Rowley, H.A.; Olney, R.K. Magnetic resonance imaging in amyotrophic lateral sclerosis. Ann. Neurol. 1988, 23, 418–420.
- Iwasaki, Y.; Kinoshita, M.; Ikeda, K.; Takamiya, K. Central nervous system magnetic resonance imaging findings in amyotrophic lateral sclerosis. Eur. Arch. Psychiatry Neurol. Sci. 1989, 239, 125–126.
- Turner, M.R.; Verstraete, E. What does imaging reveal about the pathology of amyotrophic lateral sclerosis? Curr. Neurol. Neurosci. Rep. 2015, 15, 45.
- Mills, K.R.; Murray, N.M.; Hess, C.W. Magnetic and electrical transcranial brain stimulation: Physiological mechanisms and clinical applications. Neurosurgery 1987, 20, 164–168.
- Eisen, A.A.; Shtybel, W. AAEM minimonograph #35: Clinical experience with transcranial magnetic stimulation. Muscle Nerve 1990, 13, 995–1011.
- Eisen, A.; Shytbel, W.; Murphy, K.; Hoirch, M. Cortical magnetic stimulation in amyotrophic lateral sclerosis. Muscle Nerve 1990, 13, 146–151.
- Brown, W.F.; Ebers, G.C.; Hudson, A.J.; Pringle, C.E.; Veitch, J. Motor-evoked responses in primary lateral sclerosis. Muscle Nerve 1992, 15, 626–629.
- Vucic, S.; Kiernan, M.C. Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain J. Neurol. 2006, 129, 2436–2446.
- Vucic, S.; Kiernan, M.C. Utility of transcranial magnetic stimulation in delineating amyotrophic lateral sclerosis pathophysiology. Handb. Clin. Neurol. 2013, 116, 561–575.
- Vucic, S.; Ziemann, U.; Eisen, A.; Hallett, M.; Kiernan, M.C. Transcranial magnetic stimulation and amyotrophic lateral sclerosis: Pathophysiological insights. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1161–1170.
- Vucic, S.; Howells, J.; Trevillion, L.; Kiernan, M.C. Assessment of cortical excitability using threshold tracking techniques. Muscle Nerve 2006, 33, 477–486.
- Vucic, S.; Nicholson, G.A.; Kiernan, M.C. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain J. Neurol. 2008, 131, 1540–1550.


