 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Eric J Sorin | + 3511 word(s) | 3511 | 2021-04-20 10:32:47 | | | |
| 2 | Rita Xu | Meta information modification | 3511 | 2021-04-21 04:37:50 | | |
Video Upload Options
The cholinesterase enzyme family has but two members: acetylcholinesterase (AChE) and butyrylcholinesterase (BChE). Computational efforts to better understand these enzymes, with a focus on their structural and catalytic properties and dynamics, includes docking and other Monte Carlo based calculations, as well as dynamic simulations of varying rigor and resource-demand including molecular dynamics, Langevin dynamics, quantum mechanical/molecular mechanics, and related algorithms.
1. Introduction
The cholinesterase enzyme family has but two members: acetylcholinesterase (AChE) and butyrylcholinesterase (BChE). The former’s primary biological purpose is regulating acetylcholine, a neurotransmitter, via hydrolysis at neuromuscular junctions, thus proving itself to be an essential component in the maintenance and performance of nervous systems. AChE, which has also been referred to as “true cholinesterase”, is created in muscle, nerve, and hematopoietic cells and is considered to be one of the most efficient enzymes due to its rapid rate of catalysis [1]. AChE’s plasma analog, BChE, previously referred to as “pseudocholinesterase”, is produced in the liver and, unlike AChE, has been viewed as having a much more ambiguous biological purpose, as it was long believed to be vestigial [2]. BChE has a higher concentration in plasma than AChE, is present in many vertebrates, and tolerates several mutations, which has prompted the theory that BChE evolved from AChE to be a general detoxifier while still retaining some function in the process of neurotransmission [3]. This theory seems apt given the structural similarity of their binding sites, their shared affinities for certain substrates and ligands, and their sequence homology of approximately 65% [4]. Figure 1 depicts both AChE (bottom) and BChE (top) from a gorge-centric view and after a 90° rotation.
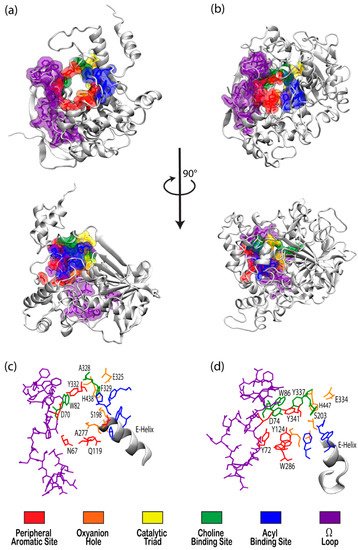
Figure 1. Visualizations of (a) butyrylcholinesterase (BChE) (PDBID 1P0I) and (b) acetylcholinesterase (AChE) (PDBID 1B41) in white ribbon mode with residues in notable binding sites shown as semi-transparent van der Waals surfaces colored according to the key. Top panels present views directly into the active site gorges with structures in the center panel rotated 90° about the vertical axis. Bottom panels present magnified views of the binding site regions of (c) BChE and (d) AChE with key residues labeled.
One difference between the two enzymes is the relative size of the binding pocket, with BChE and AChE having approximate gorge volumes of 1500 Å3 and 1300 Å3, respectively [5]. X-ray crystallography of Torpedo californica (Tc, pacific ray) AChE revealed the enzyme to have a deep hydrophobic gorge with residues that stabilize substrates in the pocket [6], as well as a bottleneck region in the active site [7] that narrows to approximately 4 Å in width [8]. Common models of AChE, including human, mouse, and Torpedo californica (TcAChE), demonstrate conserved active sites, save for a few residues that participate in ligand binding [9], and both have negative surface potentials that become more negative deeper within the gorge. This negative potential, which is high near the catalytic site at the “bottom” of the gorge, seems to have evolved to facilitate electrostatic attraction of positively charged choline substrates by both enzymes [8].
Although BChE is structurally similar to AChE, 6 of the 14 aromatic amino acids that line the active site gorge in AChE are substituted with aliphatic residues in BChE [10]. In particular, the substitution of Phe288 and Phe290 in TcAChE with the smaller Leu286 and Val288 in human BChE lead to conformational changes that result in a deeper gorge in BChE, thereby allowing BChE to interact with, and potentially hydrolize, a much wider range of substrates and inhibitors than AChE [11]. BChE is thus characterized as the promiscuous, or non-specific, bigger sibling to the smaller and much more specific AChE.
The tremendous growth and improvement in computational resources and modeling techniques over the past few decades have led to an exponential increase in computational studies of biomolecular systems. In 2003, the Protein Data Bank (PDB) launched its online presence, thereby making myriad biomolecules and complexes available for structural and computational research, and by 2016 there were 178 AChE structures available to the public, many of those including bound substrates or inhibitors of medical and pharmacological significance [9]. In fact, some of the earliest and most significant contributions of cholinesterase models were reported by Sussman et al., who performed an X-ray analysis of AChE at 2.8 resolution in 1991 [12], and Nicolet et al., who examined several crystal structures of BChE in 2003 [13]. Modeling and simulation-based studies of the cholinesterase enzymes have advanced in tandem with this growth.
The application of molecular dynamics (MD) simulations, Monte Carlo (MC) based docking calculations, and more sophisticated quantum mechanical/molecular mechanics (QM/MM) simulations have proven highly insightful in probing cholinesterase structure and activity. Notable areas of interest include the mechanisms of cholinesterase catalysis, reversible and irreversible inhibition of both enzymes to manage Alzheimer’s Disease (AD) and other ailments, BChE-specific inhibition, and the reactivation of phosphorylated cholinesterases, a process that normally follows nerve agent attacks or pesticide poisoning [14]. Figure 2a,b depict the common paths of substrate or ligand binding and phosphorylation, respectively.
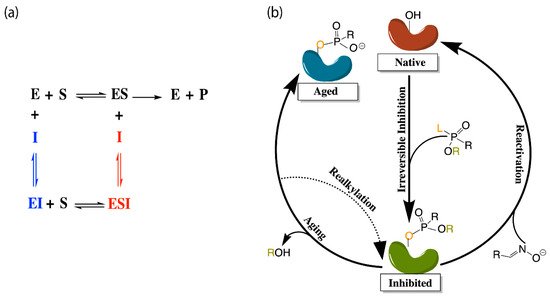
Figure 2. (a) Depiction of the enzyme catalytic mechanism (black) and reversible inhibition mechanisms including competitive inhibition (blue), uncompetitive inhibition (red), and noncompetitive inhibition (both blue and red). (b) Representation of irreversible (covalent) inhibition by organophosphorus ligands and the reactivation mechanisms to revert the aged enzyme (blue) back to the inhibited complex (green). L and R represent the first and second leaving groups of the organophosphate, respectively, following the paths of inhibition and reactivation.
Most therapeutic treatments of AD, and other illnesses to which the cholinesterases have been linked, are reversible inhibitors that form non-covalent molecular recognition (MR) complexes with cholinesterases and can leave the active site, thus existing in an equilibrium between bound and unbound states characterized by KI and/or IC50 values. The three mechanisms that reversible inhibition can follow are competitive, noncompetitive, and uncompetitive, as depicted in Figure 2a. During competitive inhibition, the substrate and inhibitor are competing for the same binding site, and the binding of a competitive inhibitor within the active site blocks entrance of the substrate, thereby hindering formation of the enzyme substrate complex. By contrast, uncompetitive inhibitors function by binding to the enzyme-substrate complex to prevent product formation, and noncompetitive inhibitors can bind to either the enzyme or the enzyme-substrate complex to regulate catalytic activity.
Many organic compounds, including organophosphates (OPs), are categorized as irreversible inhibitors, which covalently bond to residues in the gorge and thus cannot leave the active site. In the cholinesterases, OPs covalently bind to the serine residue of the catalytic triad. Irreversibly inhibited cholinesterases, however, can be reactivated via various pathways with compounds such as oximes, as illustrated in Figure 2b. Oximes reactivate an inhibited cholinesterase to its native structure by nucleophilic substitution of the phosphorylated serine. If untreated, the inhibited enzyme can undergo the dealkylation process, also known as aging, where the loss of the second leaving group produces an oxyanion on the phosphoryl group.
Aged cholinesterase is highly stable due to the strong electrostatic interactions between the oxyanion and the positively charged catalytic histidine. Known reactivators of OP-inhibited AChE, such as oximes, are ineffectual against aged AChE [15]. Though a successful attempt to reactivate aged AChE by a class of compounds called “quinone methide precursors” (QMPs) was reported by Hadad and coworkers [16], the reaction between QMPs and aged AChE was rendered too slow to be useful [17].
2. Structure and Dynamics
While understanding the structure, function, and behavior of cholinesterase binding sites seems a practical starting point for modeling studies, early researchers initially wanted to understand how substrates and inhibitors entered the active sites of these enzymes. Molecular dynamics (MD) simulations of human BChE lasting for 5 and 10 ns indicated that inhibitors can access the binding subsites in the catalytic cavity due to the highly flexible entrance (or “mouth”) of the gorge, a portion of which is formed by the flexible omega loop (Ω-loop) region, as well as the peripheral aromatic site (PAS, formerly mistakenly called the peripheral anionic site). In these simulations, the Asp70 residue in PAS showed significant deviation from the crystal structure with root-mean-square deviation (RMSD) values of 2 to 6 Å [18]. AChE has also been shown to experience such fluctuations at the gorge entrance, with a similarly flexible Ω-loop region that is thought to increase enzyme specificity by making it more difficult for large molecules to enter the AChE gorge without hindering the productivity of the enzyme [19].
Apart from the fluctuations at the mouth of the gorge, AChE also experiences what is known as bottleneck fluctuations, with the bottleneck being the narrowest part of the gorge. These fluctuations, which have come to be known as the “breathing” of the enzyme, can help inhibitors or substrates move from the surface to deeper regions of the gorge [20]. Cheng et al. recently defined this breathing by monitoring the varying distance between the Cε2 atom of Phe330 (CBS) and the OH atom of Tyr121 (PAS) in TcAChE, which suggested that a number of subdomains within the enzyme, particularly the Ω-loop, contribute to modulating the size of the gorge bottleneck [21]. A comprehensive comparison between 47 crystal structures of AChE (in its native form and in complex with small molecules), as well as a 20 ns simulation of TcAChE, provided by Sussman and coworkers, suggested that the 14 aromatic residues lining the AChE gorge and creating over half of the gorge surface area contribute greatly to the overall flexibility of the enzyme, the observed bottleneck breathing motions, and the resulting ability to perform its catalytic function [22].
Some of these aromatic residues play significant roles in primary subsites within the cholinesterase gorges, such as the catalytic active site (CAS) and the peripheral aromatic site (PAS) [22]. While these sites, which are addressed in more detail below, are integral to cholinergic activity, cholinesterases do not only perform cholinergic functions. As discussed by Chinnadurai et al., the aryl acylamidase activity (AAA) of AChE, which also involves hydrolysis, only requires the CAS and does not interact with the PAS at all [23]. This has prompted the theory that AAA substrates enter from a side-door into the enzyme, rather than via the mouth of the gorge and, indeed, researchers have suggested that there are a number of doors through which substrates can enter the gorge including a back door [24], an Ω-loop door, and the suggested side door [23]. It was suggested that the side door may open more frequently than the other doors to mediate AAA activity, and MD simulations of side door probing emphasize the importance of hydrophobic interactions, hydrogen bonding, and water mediated interactions (“water bridges”) in moving the substrate towards the CAS [23]. To be sure, simulations of AChE in explicit solvent sans substrate [25], as well as analyses of TcAChE crystal structures in its native and several inhibited forms [26], have underscored the importance of the presence of molecular water in enzyme structure.
At higher concentrations, dimerization and the further dimerization of dimers to form tetramers is known to affect the structure and function of cholinesterase enzymes [27]. While MD simulations suggested that two of the four binding sites in tetramerized cholinesterases are sterically blocked, thereby becoming less active [28], as reflected by a 15% decrease in catalytic activity [29], a recently elucidated CryoEM structure of hBChE shows distinct structural variance from the simulated tetramer, with the active site gorges being fully solvent accessible [27]. Tetramerization, however, increases the half-life of the enzymes, which is a desirable result when cholinesterases, and BChE in particular, are being used to counteract drug overdoses. For example, the addition of proline-rich attachment domains (PRADs) to BChE increases tetramer stability, leading to an extended circulation time [30]. Interestingly, glycosylated models of BChE increase the enzyme’s flexibility and half-life without hindering its ability to bind to glycans, which cannot be said of all therapeutic protein targets [31]. On the other hand, cholinesterase phosphonylation, or the irreversible binding of organophosphate to the active site, which will be discussed in more detail in the organophosphate inhibition section below, severely restricts the flexibility of both AChE and BChE, as reported by Bennion et al. [32]. Experimental findings of AChE covalently bound to the nerve agent soman agree that OP-poisoned AChE is significantly stiffer [33].
2.1. Important Binding Sites
There has been substantial past effort to study the sites responsible for molecular recognition (MR) and binding affinity within the active site gorge of both enzymes, and it is clear that those binding sites, specific chemical subsites within each active site gorge, mirror each other and perform similar functions respective to each enzyme. While it is important to note that these binding sites have been examined experimentally with X-ray and kinetic studies, including a recent study by Rosenberry et al. [34], the present review focuses on the unique perspective provided by computational investigations. As expected, one of the most important sites for both cholinesterases is the catalytic active site (CAS), and the peripheral aromatic site (PAS) also plays an indispensable role in cholinesterase or ligand binding, while the Ω-loop (OML), acyl binding site (ABS), and oxyanion hole (OAH) sites are more essential in contributing to binding affinity and complex stability. Alvarado et al. have provided a method of succinct graphical tabulation of BChE-ligand contacts and interactions, referred to as contact tables, that include these five sites and additional protein residues of interest [35], as discussed below. A detailed analysis of these binding sites is provided here in the same order that they are encountered by substrates and inhibitors upon entering and moving into the gorge.
2.1.1. Peripheral Aromatic Site
The peripheral aromatic site (PAS, red in Figure 1) is located near the mouth of gorge [36] and plays a prominent role in substrate and ligand binding [37]. For decades, peer-reviewed studies have used PAS to denote the “peripheral anionic site”. In recent years, however, the aromatic properties of this binding site that are vital to cholinesterase function have driven the community to instead refer to this region as the “peripheral aromatic site”. Important amino acids in the PAS of AChE include serine, tyrosine, aspartic acid, and tryptophan [36][38], while notable PAS residues in BChE include asparagine, aspartic acid, glutamine, serine, and tyrosine [35], highlighting the polar, negatively charged, and electron-rich nature of residues in this site. As previously mentioned, one distinction between the cholinesterases is the aromatic nature of the residues surrounding the PAS in AChE, which is more aliphatic in BChE [10]. The PAS makes contact with many loops and secondary structural elements at the surface of the protein, including the Ω-loop, which contributes to the needed flexibility discussed above. Although steric and electrostatic interactions may slow the catalytic efficiency of AChE, the PAS is valuable for trafficking ligands into the gorge [39], particularly positively charged species such as cholines. MD simulations have emphasized the importance of cation-π interactions, which stabilize the ligand at the rim of the gorge entrance prior to entering the gorge [40], and it has been proposed that non-cholinergic activity of the PAS could include the deposition of amyloids, adhesion to cells, and outgrowth of neurites [41].
2.1.2. Acyl and Choline Binding Sites
Once a ligand has entered the gorge, the acyl and choline binding sites (ABS and CBS, shown as blue and green in Figure 1, respectively), which are located near the catalytic triad, assist in positioning the ligand for catalysis. The ABS and CBS are hydrophobic regions composed primarily of tryptophan, tyrosine, and phenylalanine in human AChE. Tyr337 in the choline binding site of AChE is replaced by Ala328 in that of BChE; Phe295 and Phe297 in the acyl binding site of AChE are replaced by Leu286 and Val288, respectively, in BChE. The replacement of aromatic residues in the ABS and CBS of BChE enable it to bind larger substrates and inhibitors than AChE [42]. In addition, the ABS and CBS are largely responsible for the specificity of these enzymes and are thus primary targets studied when synthesizing inhibitors such as imidazole or pyridine derivatives [43].
2.1.3. Catalytic Active Site
The catalytic active site (CAS, yellow in Figure 1) is located approximately 20 Å deep at the bottom of both the AChE and BChE gorges [12][24] and is made up of serine, glutamic acid, and histidine residues, prompting the name “catalytic triad” [24][35]. The CAS is surrounded by numerous aromatic and acidic residues [44] and is observed to engage in shorter hydrogen bonds in crystal and NMR structures than observed in simulation [45][46]. More importantly, the CAS is highly conserved [44], emphasizing historically vital biological roles of these enzymes and their cholinergic activities. QM/MM simulations at the MP2(6-31 + G*) level reveal a potential energy barrier of 10.5 kcal/mol, which agrees with experimental data [47], and MD simulations of AChE bound to acetylcholine (ACh) show that ACh stabilizes the CAS and improves the binding ability of the peripheral aromatic site [48].
It has been postulated that a back-door exists in AChE, just behind the CAS and controlled by a tryptophan residue, which was theorized after a single water molecule exited the active site gorge from a direction opposite that of the gorge entrance in an MD simulation [49]. This “back door” was later thought to open three to four Å wide such that catalysis products could exit the gorge of the enzyme without blocking the gorge entrance, and thus contributing to a high catalytic rate [50]. Aromatic residues surrounding the CAS histidine also largely influence the productivity and efficiency of the enzyme [51], which decreased approximately 600-fold when disrupted or replaced by aliphatic side chains [52]. More recently, Xu et al. used MD to study TcAChE and, from 27 of their 40 trajectories, observed thiocholine to frequently exit the active site via a back-door created by cooperative motions of CAS residue Trp86 along with Val132 and Gly448 [53], for which previous experimental support was noted [54][55].
Indeed, other mutations in or near the CAS are known to have effects on the structure, and subsequent function, of the enzyme. This research has naturally focused on, and is more applicable to, BChE due to its much greater natural affinity for mutations than AChE [3]. For example, prolonged use of muscle relaxers led to the discovery of the “silent phenotype” in which an alanine is mutated to a valine near the CAS of BChE. This mutation was studied in silico and observed to severely disrupt interactions between the histidine and serine in the catalytic triad [56], leading to a dysfunctional CAS, regardless of the inhibitor, and increases in the volume of the enzyme, indicating that this may be a pre-denaturation state [57].
The mutation of a nearby alanine in silico to cystine in wild-type (WT) BChE causes the histidine in the catalytic triad to flip, an event that is largely guided by local water molecules [58]. This man-made mutation, while possibly slowing the speed of binding, ultimately still allows for substrate binding to the active site; the naturally occurring mutation of that alanine to aspartic acid, however, is claimed to be catalytically inactive due to strong disruptive interactions between aspartic acid and the CAS histidine residue [59]. Both mutations showcase the possible hysteretic behavior of BChE, or its reliance on past-states, which can likely be attributed to its toxicological and pharmaceutical functions [58][59]. For instance, a man-made mutant of BChE was recently modeled and examined by Masson and coworkers, using QM/MM and Markov state analysis, and was determined to be a template for future investigations into organophosphate hydrolase functions [60].
2.1.4. Oxyanion Hole
The oxyanion hole (OAH, orange in Figure 1), made of two glycines and one alanine [35][47], is generally a two-pronged site in many proteases and hydrolases; in the case of AChE, and subsequently BChE, the OAH is a three-pronged hole [61]. Early MD simulations of AChE phosphonylation suggested that the OAH exerts a pulling force on leaving groups during the alkylation step [45], and the OAH is known to lower the energy barrier for ACh hydrolysis in both cholinesterases [62]. QM/MM simulations exhibited consistent, tight hydrogen bonding between the OAH and the carbonyl carbon of the substrate, suggesting that the OAH facilitates stabilizing interactions in intermediate and transition states [61].
2.1.5. Ω-loop (Omega Loop)
The Ω-loop (OML, purple in Figure 1), consisting of a series of nearly 30 residues, is located along one side of the active site gorge wall. In the presence of a substrate or inhibitor, the Ω-loop plays an important role in modulating enzyme “breathing”, regulating the size of the gorge, and thereby enabling the passage of the ligand to the active site [21]. The OML undergoes conformational changes, such as gorge enlargement, through torsional motion and segmental fluctuations [63][64]. Unregulated motions and decreased electrostatic interactions, however, can significantly decrease the binding affinity of these enzymes, such as the case of the atypical mutation from Asp70 to Gly70 in the OML of BChE, which Masson et al. reported could increase the Km values 10- to 100-fold [65]. Moreover, the Ω-loop is speculated to facilitate an alternative entrance, the proposed side-door model noted above, to the active site of the enzyme; MD simulations performed by Wiesner et al. suggested that protonation of the AChE active site leads to conformational changes within the Ω-loop at Asn87 and Glu84 that result in the opening of this side door [66]. A similar observation was recently reported by the Rydzewski laboratory for TcAChE, where opening and closing of this side door due to the displacement of the OML favored alternative dissociate routes of the substrate and inhibitor [67]. To put these computational results into perspective, a number of experimental investigations into the omega loop and backdoor of AChE from various species have suggested that the back door opens in some cases [50][68][69], but also that this opening is likely not relevant functionally [70].
References
- Taylor, P.; Camp, S.; Radić, Z. Acetylcholinesterase. In Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Oxford, UK, 2009.
- John, H.; Balszuweit, F.; Kehe, K.; Worek, F.; Thiermann, H. Chapter 50—Toxicokinetics of Chemical Warfare Agents: Nerve Agents and Vesicants. In Handbook of Toxicology of Chemical Warfare Agents; Gupta, R.C., Ed.; Academic Press: San Diego, CA, USA, 2009.
- Johnson, G.; Moore, S.W. Why has butyrylcholinesterase been retained? Structural and functional diversification in a duplicated gene. Neurochem. Int. 2012, 61, 783–797.
- Ortiz, J.E.; Pigni, N.B.; Andujar, S.A.; Roitman, G.; Suvire, F.D.; Enriz, R.D.; Tapia, A.; Bastida, J.; Feresin, G.E. Alkaloids from Hippeastrum argentinum and Their Cholinesterase-Inhibitory Activities: An in Vitro and in Silico Study. J. Nat. Prod. 2016, 79, 1241–1248.
- Masson, P.; Lushchekina, S.; Schopfer, L.M.; Lockridge, O. Effects of viscosity and osmotic stress on the reaction of human butyrylcholinesterase with cresyl saligenin phosphate, a toxicant related to aerotoxic syndrome: Kinetic and molecular dynamics studies. Biochem. J. 2013, 454, 387–399.
- Lee, S.; Barron, M.G. A mechanism-based 3D-QSAR approach for classification and prediction of acetylcholinesterase inhibitory potency of organophosphate and carbamate analogs. J. Comput. Aided Mol. Des. 2016, 30, 347–363.
- Kharlamova, A.D.; Lushchekina, S.V.; Petrov, K.A.; Kots, E.D.; Nachon, F.; Villard-Wandhammer, M.; Zueva, I.V.; Krejci, E.; Reznik, V.S.; Zobov, V.V.; et al. Slow-binding inhibition of acetylcholinesterase by an alkylammonium derivative of 6-methyluracil: Mechanism and possible advantages for myasthenia gravis treatment. Biochem. J. 2016, 473, 1225–1236.
- Felder, C.E.; Botti, S.A.; Lifson, S.; Silman, I.; Sussman, J.L. External and internal electrostatic potentials of cholinesterase models. J. Mol. Graph. Model. 1997, 15, 318–327.
- Ochoa, R.; Rodriguez, C.A.; Zuluaga, A.F. Perspectives for the structure-based design of acetylcholinesterase reactivators. J. Mol. Graph. Model. 2016, 68, 176–183.
- Saxena, A.; Redman, A.M.G.; Jiang, X.; Lockridge, O.; Doctor, B.P. Differences in Active Site Gorge Dimensions of Cholinesterases Revealed by Binding of Inhibitors to Human Butyrylcholinesterase. Biochemistry 1997, 36, 14642–14651.
- Ul-Haq, Z.; Khan, W.; Kalsoom, S.; Ansari, F.L. In silico modeling of the specific inhibitory potential of thiophene-2,3-dihydro-1,5-benzothiazepine against BChE in the formation of beta-amyloid plaques associated with Alzheimer’s disease. Theor. Biol. Med. Model. 2010, 7, 26.
- Sussman, J.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879.
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal Structure of Human Butyrylcholinesterase and of Its Complexes with Substrate and Products. J. Biol. Chem. 2003, 278, 41141–41147.
- Dvir, H.; Silman, I.; Harel, M.; Rosenberry, T.L.; Sussman, J.L. Acetylcholinesterase: From 3D structure to function. Chem. Biol. Interact. 2010, 187, 10–22.
- Zhuang, Q.G.; Young, A.; Callam, C.S.; McElroy, C.A.; Ekici, O.D.; Yoder, R.J.; Hadad, C.M. Efforts toward treatments against aging of organophosphorus-inhibited acetylcholinesterase. Ann. N. Y. Acad. Sci. 2016, 1374, 94–104.
- Zhuang, Q.G.; Franjesevic, A.J.; Corrigan, T.S.; Coldren, W.H.; Dicken, R.; Sillart, S.; DeYong, A.; Yoshino, N.; Smith, J.; Fabry, S.; et al. Demonstration of In Vitro Resurrection of Aged Acetylcholinesterase after Exposure to Organophosphorus Chemical Nerve Agents. J. Med. Chem. 2018, 61, 7034–7042.
- Quinn, D.M. Resurrection Biology: Aged Acetylcholinesterase Brought Back to Life. J. Med. Chem. 2018, 61, 7032–7033.
- Suárez, D.; Field, M.J. Molecular dynamics simulations of human butyrylcholinesterase. Proteins Struct. Funct. Bioinform. 2005, 59, 104–117.
- Zhou, H.-X.; Wlodek, S.T.; McCammon, J.A. Conformation gating as a mechanism for enzyme specificity. Proc. Natl. Acad. Sci. USA 1998, 95, 9280–9283.
- Shen, T.; Tai, K.; Henchman, R.H.; McCammon, J.A. Molecular Dynamics of Acetylcholinesterase. Acc. Chem. Res. 2002, 35, 332–340.
- Cheng, S.M.; Song, W.L.; Yuan, X.J.; Xu, Y.C. Gorge Motions of Acetylcholinesterase Revealed by Microsecond Molecular Dynamics Simulations. Sci. Rep. 2017, 7, 3219.
- Xu, Y.; Colletier, J.-P.; Weik, M.; Jiang, H.; Moult, J.; Silman, I.; Sussman, J.L. Flexibility of Aromatic Residues in the Active-Site Gorge of Acetylcholinesterase: X-ray versus Molecular Dynamics. Biophys. J. 2008, 95, 2500–2511.
- Chinnadurai, R.K.; Saravanaraman, P.; Boopathy, R. Understanding the molecular mechanism of aryl acylamidase activity of acetylcholinesterase—An in silico study. Arch. Biochem. Biophys. 2015, 580, 1–13.
- Axelsen, P.H.; Harel, M.; Silman, I.; Sussman, J.L. Structure and dynamics of the active site gorge of acetylcholinesterase: Synergistic use of molecular dynamics simulation and X-ray crystallography. Protein Sci. 1994, 3, 188–197.
- Henchman, R.H.; McCammon, J.A. Structural and dynamic properties of water around acetylcholinesterase. Protein Sci. 2002, 11, 2080–2090.
- Koellner, G.; Kryger, G.; Millard, C.B.; Silman, I.; Sussman, J.L.; Steiner, T. Active-site gorge and buried water molecules in crystal structures of acetylcholinesterase from Torpedo californica. J. Mol. Biol. 2000, 296, 713–735.
- Leung, M.R.; van Bezouwen, L.S.; Schopfer, L.M.; Sussman, J.L.; Silman, I.; Lockridge, O.; Zeev-Ben-Mordehaia, T. Cryo-EM structure of the native butyrylcholinesterase tetramer reveals a dimer of dimers stabilized by a superhelical assembly. Proc. Natl. Acad. Sci. USA 2018, 115, 13270–13275.
- Fang, L.; Pan, Y.; Muzyka, J.L.; Zhan, C.-G. Active Site Gating and Substrate Specificity of Butyrylcholinesterase and Acetylcholinesterase: Insights from Molecular Dynamics Simulations. J. Phys. Chem. B 2011, 115, 8797–8805.
- Gorfe, A.A.; Lu, B.Z.; Yu, Z.Y.; McCammon, J.A. Enzymatic Activity versus Structural Dynamics: The Case of Acetylcholinesterase Tetramer. Biophys. J. 2009, 97, 897–905.
- Pan, Y.; Muzyka, J.L.; Zhan, C.-G. Model of Human Butyrylcholinesterase Tetramer by Homology Modeling and Dynamics Simulation. J. Phys. Chem. B 2009, 113, 6543–6552.
- Fang, L.; Zheng, F.; Zhan, C.-G. A model of glycosylated human butyrylcholinesterase. Mol. Biosyst. 2014, 10, 348–354.
- Bennion, B.J.; Essiz, S.G.; Lau, E.Y.; Fattebert, J.-L.; Emigh, A.; Lightstone, F.C. A wrench in the works of human acetylcholinesterase: Soman induced conformational changes revealed by molecular dynamics simulations. PLoS ONE 2015, 10, e0121092.
- Peters, J.; Martinez, N.; Trovaslet, M.; Scannapieco, K.; Koza, M.M.; Masson, P.; Nachon, F. Dynamics of human acetylcholinesterase bound to non-covalent and covalent inhibitors shedding light on changes to the water network structure. Phys. Chem. Chem. Phys. 2016, 18, 12992–13001.
- Rosenberry, T.L.; Brazzolotto, X.; Macdonald, I.R.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F. Comparison of the Binding of Reversible Inhibitors to Human Butyrylcholinesterase and Acetylcholinesterase: A Crystallographic, Kinetic and Calorimetric Study. Molecules 2017, 22, 2098.
- Alvarado, W.; Bremer, P.L.; Choy, A.; Dinh, H.N.; Eung, A.; Gonzalez, J.; Ly, P.; Tran, T.; Nakayama, K.; Schwans, J.P.; et al. Understanding the enzyme-ligand complex: Insights from all-atom simulations of butyrylcholinesterase inhibition. J. Biomol. Struct. Dyn. 2019, 38, 1028–1041.
- Bourne, Y.; Taylor, P.; Bougis, P.E.; Marchot, P. Crystal structure of mouse acetylcholinesterase—A peripheral site-occluding loop in a tetrameric assembly. J. Biol. Chem. 1999, 274, 2963–2970.
- Campiani, G.; Fattorusso, C.; Butini, S.; Gaeta, A.; Agnusdei, M.; Gemma, S.; Persico, M.; Catalanotti, B.; Savini, L.; Nacci, V.; et al. Development of molecular probes for the identification of extra interaction sites in the mid-gorge and peripheral sites of butyrylcholinesterase (BuChE). Rational design of novel, selective, and highly potent BuChE inhibitors. J. Med. Chem. 2005, 48, 1919–1929.
- Khan, M.T.H. Molecular interactions of cholinesterases inhibitors using in silico methods: Current status and future prospects. New Biotechnol. 2009, 25, 331–346.
- Roca, C.; Requena, C.; Sebastian-Perez, V.; Malhotra, S.; Radoux, C.; Perez, C.; Martinez, A.; Antonio Paez, J.; Blundell, T.L.; Campillo, N.E. Identification of new allosteric sites and modulators of AChE through computational and experimental tools. J. Enzym. Inhib. Med. Chem. 2018, 33, 1034–1047.
- Branduardi, D.; Gervasio, F.L.; Cavalli, A.; Recanatini, M.; Parrinello, M. The role of the peripheral anionic site and cation-pi interactions in the ligand penetration of the human AChE gorge. J. Am. Chem. Soc. 2005, 127, 9147–9155.
- Johnson, G.; Moore, S. The peripheral anionic site of acetylcholinesterase: Structure, functions and potential role in rational drug design. Curr. Pharm. Des. 2006, 12, 217–225.
- Dighe, S.N.; Deora, G.S.; De la Mora, E.; Nachon, F.; Chan, S.; Parat, M.O.; Brazzolotto, X.; Ross, B.P. Discovery and Structure-Activity Relationships of a Highly Selective Butyrylcholinesterase Inhibitor by Structure-Based Virtual Screening. J. Med. Chem. 2016, 59, 7683–7689.
- Kwong, H.C.; Chidan Kumar, C.S.; Mah, S.H.; Mah, Y.L.; Chia, T.S.; Quah, C.K.; Lim, G.K.; Chandraju, S. Crystal Correlation Of Heterocyclic Imidazo[1,2-a]pyridine Analogues and Their Anticholinesterase Potential Evaluation. Sci. Rep. 2019, 9, 926.
- Kumar, J.; Gill, A.; Shaikh, M.; Singh, A.; Shandilya, A.; Jameel, E.; Sharma, N.; Mrinal, N.; Hoda, N.; Jayaram, B. Pyrimidine-Triazolopyrimidine and Pyrimidine-Pyridine Hybrids as Potential Acetylcholinesterase Inhibitors for Alzheimer’s Disease. ChemistrySelect 2018, 3, 736–747.
- Bencsura, A.; Enyedy, I.Y.; Kovach, I.M. Probing the active site of acetylcholinesterase by molecular dynamics of its phosphonate ester adducts. J. Am. Chem. Soc. 1996, 118, 8531–8541.
- Viragh, C.; Harris, T.K.; Reddy, P.M.; Massiah, M.A.; Mildvan, A.S.; Kovach, I.M. NMR Evidence for a Short, Strong Hydrogen Bond at the Active Site of a Cholinesterase. Biochemistry 2000, 39, 16200–16205.
- Zhang, Y.K.; Kua, J.; McCammon, J.A. Role of the catalytic triad and oxyanion hole in acetylcholinesterase catalysis: An ab initio QM/MM study. J. Am. Chem. Soc. 2002, 124, 10572–10577.
- Kua, J.; Zhang, Y.; McCammon, J.A. Studying Enzyme Binding Specificity in Acetylcholinesterase Using a Combined Molecular Dynamics and Multiple Docking Approach. J. Am. Chem. Soc. 2002, 124, 8260–8267.
- Gilson, M.K.; Straatsma, T.P.; McCammon, J.A.; Ripoli, D.R.; Faerman, C.H.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Open “back door” in a molecular dynamics simulation of acetylcholinesterase. Science 1994, 263, 1276–1278.
- Sanson, B.; Colletier, J.-P.; Xu, Y.; Lang, P.T.; Jiang, H.; Silman, I.; Sussman, J.L.; Weik, M. Backdoor opening mechanism in acetylcholinesterase based on X-ray crystallography and molecular dynamics simulations. Protein Sci. 2011, 20, 1114–1118.
- Kaplan, D.; Barak, D.; Ordentlich, A.; Kronman, C.; Velan, B.; Shafferman, A. Is aromaticity essential for trapping the catalytic histidine 447 in human acetylcholinesterase? Biochemistry 2004, 43, 3129–3136.
- Barak, D.; Kaplan, D.; Ordentlich, A.; Ariel, N.; Velan, B.; Shafferman, A. The aromatic “trapping” of the catalytic histidine is essential for efficient catalysis in acetylcholinesterase. Biochemistry 2002, 41, 8245–8252.
- Xu, Y.; Colletier, J.-P.; Weik, M.; Qin, G.; Jiang, H.; Silman, I.; Sussman, J.L. Long route or shortcut? A molecular dynamics study of traffic of thiocholine within the active-site gorge of acetylcholinesterase. Biophys. J. 2010, 99, 4003–4011.
- Colletier, J.; Royant, A.; Specht, A.; Sanson, B.; Nachon, F.; Masson, P.; Zaccai, G.; Sussman, J.; Goeldner, M.; Silman, I.; et al. Use of a ‘caged’ analogue to study the traffic of choline within acetylcholinesterase by kinetic crystallography. Acta Crystallogr. D 2007, 63, 1115–1128.
- Colletier, J.-P.; Bourgeois, D.; Sanson, B.; Fournier, D.; Sussman, J.L.; Silman, I.; Weik, M. Shoot-and-Trap: Use of specific X-ray damage to study structural protein dynamics by temperature-controlled cryo-crystallography. Proc. Natl. Acad. Sci. USA 2008, 105, 11742–11747.
- Delacour, H.; Lushchekina, S.; Mabboux, I.; Ceppa, F.; Masson, P.; Schopfer, L.M.; Lockridge, O. Characterization of a novel butyrylcholinesterase point mutation (p.Ala34Val), “silent” with mivacurium. Biochem. Pharmacol. 2014, 92, 476–483.
- Delacour, H.; Lushchekina, S.; Mabboux, I.; Bousquet, A.; Ceppa, F.; Schopfer, L.M.; Lockridge, O.; Masson, P. Characterization of a Novel BCHE “Silent’’ Allele: Point Mutation (p. Val204Asp) Causes Loss of Activity and Prolonged Apnea with Suxamethonium. PLoS ONE 2014, 9, e101552.
- Lushchekina, S.V.; Nemukhin, A.V.; Varfolomeev, S.D.; Masson, P. Molecular Modeling Evidence for His438 Flip in the Mechanism of Butyrylcholinesterase Hysteretic Behavior. J. Mol. Neurosci. 2014, 52, 434–445.
- Lushchekina, S.; Nemukhin, A.; Varfolomeev, S.; Masson, P. Understanding the non-catalytic behavior of human butyrylcholinesterase silent variants: Comparison of wild-type enzyme, catalytically active Ala328Cys mutant, and silent Ala328Asp variant. Chem. Biol. Interact. 2016, 259, 223–232.
- Grigorenko, B.L.; Novichkova, D.A.; Lushchekina, S.V.; Zueva, I.V.; Schopfer, L.M.; Nemukhin, A.V.; Varfolomeev, S.D.; Lockridge, O.; Masson, P. Computer-designed active human butyrylcholinesterase double mutant with a new catalytic triad. Chem. Biol. Interact. 2019, 306, 138–146.
- Zheng, F.; Yang, W.C.; Xue, L.; Hou, S.R.; Liu, J.J.; Zhan, C.G. Design of High-Activity Mutants of Human Butyrylcholinesterase against (-)-Cocaine: Structural and Energetic Factors Affecting the Catalytic Efficiency. Biochemistry 2010, 49, 9113–9119.
- Gao, D.; Zhan, C.-G. Modeling Effects of Oxyanion Hole on the Ester Hydrolysis Catalyzed by Human Cholinesterases. J. Phys. Chem. B 2005, 109, 23070–23076.
- Shi, J.X.; Tai, K.; McCammon, J.A.; Taylor, P.; Johnson, D.A. Nanosecond dynamics of the mouse acetylcholinesterase Cys(69)-Cys(96) omega loop. J. Biol. Chem. 2003, 278, 30905–30911.
- Shi, J.; Boyd, A.E.; Radic, Z.R.; Taylor, P. Reversibly Bound and Covalently Attached Ligands Induce Conformational Changes in the Omega Loop, Cys69–Cys96, of Mouse Acetylcholinesterase*. J. Biol. Chem. 2001, 276, 42196–42204.
- Masson, P.; Legrand, P.; Bartels, C.F.; Froment, M.T.; Schopfer, L.M.; Lockridge, O. Role of aspartate 70 and tryptophan 82 in binding of succinyldithiocholine to human butyrylcholinesterase. Biochemistry 1997, 36, 2266–2277.
- Wiesner, J.; Kriz, Z.; Kuca, K.; Jun, D.; Koca, J. Influence of the Acetylcholinesterase Active Site Protonation on Omega Loop and Active Site Dynamics. J. Biomol. Struct. Dyn. 2010, 28, 393–403.
- Rydzewski, J.; Jakubowski, R.; Nowak, W.; Grubmuller, H. Kinetics of Huperzine A Dissociation from Acetylcholinesterase via Multiple Unbinding Pathways. J. Chem. Theory Comput. 2018, 14, 2843–2851.
- Bourne, Y.; Renault, L.; Marchot, P. Crystal Structure of Snake Venom Acetylcholinesterase in Complex with Inhibitory Antibody Fragment Fab410 Bound at the Peripheral Site evidence for open and closed states of a back door channel. J. Biol. Chem. 2015, 290, 1522–1535.
- Nachon, F.; Rosenberry, T.L.; Silman, I.; Sussman, J.L. A Second Look at the Crystal Structures of Drosophila melanogaster Acetylcholinesterase in Complex with Tacrine Derivatives Provides Insights Concerning Catalytic Intermediates and the Design of Specific Insecticides. Molecules 2020, 25, 1198.
- Nachon, F.; Stojan, J.; Fournier, D. Insights into substrate and product traffic in the Drosophila melanogaster acetylcholinesterase active site gorge by enlarging a back channel. FEBS J. 2008, 275, 2659–2664.

