 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Xiaotian Zhong | + 4949 word(s) | 4949 | 2021-04-06 08:42:23 | | | |
| 2 | Vivi Li | Meta information modification | 4949 | 2021-04-21 03:23:45 | | |
Video Upload Options
Multispecific biotherapeutic drugs are protein-based therapeutic molecules that can engage multiple drug-target binding interfaces concurrently.
1. Introduction
Biopharmaceutical innovation has entered a new age of prosperity with record-breaking approvals for new molecular entities and biologics by the US Food and Drug Administration for the past few years [1]. Along with small-molecule entities, biotherapeutics drugs such as antibodies make up a significant portion of these approved new medicines. Novel therapeutic modalities such as RNA-based therapies, cell-based therapies, and gene-therapies are also available to patients who desperately need these medicines. The rise of this innovation wave builds on the rational drug design principle harnessing the power of genomics, proteomics, and metabolomics. The conventional mono-targeting drug model developed in the 1970s has worked exceptionally well on those “low-hanging fruit” targets [2][3], yet its effectiveness has been challenged by the growing complexity of yet-to-be addressed human diseases. Disappointing clinical studies and significant unmet medical needs remain abundant. As a response to these arduous tasks, the multispecific concept that targets two or more entities has been gradually picked up by the industry for the past decade. The multispecific biotherapeutics drug candidates have made up a significant portion of clinical and preclinical pipelines of large and small biopharmaceutical companies [4][5][6]. Developing multispecific drugs-based therapeutic agents have the potential not only to engage multiple therapeutic targets in disease pathways, but also to employ multiple modes of action and biological effectors. The power of antibody technology, the marriage between protein and chemistry (antibody drug conjugation), and the advancements of recombinant DNA technology, have put the biotherapeutics drugs into the forefront of the multispecific innovation wave.
The concept of artificially combining two distinct antigen binding paratopes targeting either the same or different antigens into one single molecule was first demonstrated by Nisonoff and Rivers sixty years ago [7]. A natural bispecific antibody formed by chain exchange of two IgG4 antibodies was later observed [8]. Current multispecific biotherapeutics can be roughly classified into two major categories, the non-obligate and the obligate multispecific [3][4][5][9][10][11]. Non-obligate multispecific biotherapeutics are exemplified by recombinant antibody-based molecules that bind two or more targets involved in independent signaling pathways (e.g., cytokines) thereby modulating these pathways simultaneously. Their efficacies could be similar to those of a mixture of separate antibodies, but the non-obligate multispecific biotherapeutics have the regulatory advantages of a single agent. In contrast, obligate multispecific biotherapeutics need the multiple binding specificities to be included within a single molecule to achieve expected efficacies. These molecules include immune-cell engagers (bringing immune cells to tumor cells to proximity for killing), antibody-drug conjugates (delivering small molecule drugs to target location via antibody), tetherbodies (enriching target molecules (cytokines or immunotoxin) into specific tissue/cellular locations), and biologic matchmaker drugs (close proximity to an action partner, e.g., emicizumab that brings substate Factor X (FX) to protease Factor IXa (FIXa) for cleavage).
For the obvious advantages in therapeutics application, taking the seemingly simple idea of multispecific-targeting into therapeutic reality has been challenging. The T-cell recruiting bispecific antibodies that killed tumor cells were first reported in 1985 [12][13]. Subsequently in the 1990s, a number of clinical trials were initiated testing the bispecific antibodies, yet with disappointing results the trials were discontinued [11]. Rapid and uncontrolled T-cell mediated cytokine release at very low dose of the drug hindered further attempts in development, along with issues in production and molecular instability. The final approval of blinatumomab for acute lymphocytic leukemia in 2014 marked the proof of principle of the T-cell engagement concept, though blinatumomab’s initial phase I trials (short-term intravenous infusion (IV), over 2–4 h at doses in the range of 0.75–13 µg/m2 up to three times per week) were terminated early due to instances of severe cytokine-release syndrome, neurotoxicity and lack of efficacy [5][14]. Only after the mode of administration was switched to long-term continuous IV infusion (for 4 or 8 weeks at doses in the range of 0.5–90 µg/m2 per day), impressive clinical results were finally achieved and sparked the evolution of the concept of bispecific T-cell engagers [4][5].
2. Major Classes of Multispecific Biotherapeutic Drugs
By definition, multispecific biotherapeutic drugs are protein-based therapeutic molecules that can engage multiple drug-target binding interfaces concurrently. The modalities of these biomolecules can be antibodies, different formats of antigen-binding fragments, small scaffold protein domains, peptides, enzymatic domains, protein receptors, chemical molecules, or oligonucleotides like anti-sense RNA or small interference RNA (siRNA). These biological entities are covalently linked together to form a single molecule to achieve multiple therapeutic modes of action. For the protein components of multispecific biotherapeutics, they are typically generated through recombinant DNA technology. For entities like chemical molecules, oligonucleotides, and some peptides, they are covalently conjugated to the protein moieties through in vitro manipulation.
Antigen-binding moieties are the dominant components for multispecific biotherapeutics as they are the main building blocks for further genetic fusion or chemical conjugation. The molecular designs for the antigen-binding moieties have been extensively reviewed [9][10], with around 100 different formats developed over the past two decades. These formats consist of various binding modules arranged in different domain repeats and fusion orders. As summarized in Table 1, the binding modules typically include fragments of antibody (Fabs), single-chain variable fragment (scFv), diabodies, single-domain antibodies (VHH from llama or camel, VNAR from shark), synthetic peptides, receptor domains, enzyme domains, CH2-based nanoantibodies or non-antibody scaffolds (Affibody, Fynomers, Monobodies, DARPins, Knottins, VLRs) [9][10][15][16][17]. The connections between these modules are either amino acid linkers encoded by DNA or chemical linkers synthesized in vitro.
One important criterion for multispecific polypeptides is the presence or absence of the antibody’s “fragment crystallizable region” (Fc) portion which plays a critical role in effector functions and FcRn-mediated recycling [18][19]. Based on the heterodimerization of Fc, multispecific molecules can be further classified into symmetric [9][10][20][21] or asymmetric architectures [4][22][23]. Symmetric multispecific biotherapeutics are homodimers with identical binding modules in each Fc fusion monomer, whereas asymmetric multispecific biotherapeutics are heterodimers with different binding modules in either of the Fc fusion monomers.
One unique problem posed for Fc-containing antibody-like multispecific biotherapeutics is the “chain-pairing” issue (detailed discussions are seen in several excellent reviews [4][9][10][22]), due to their need for the correct assembly of two antibody-like heavy chain (HC) polypeptides, and two or more antibody-like light chain (LC) polypeptides into the final multispecific molecules. A number of technologies have been developed to overcome this “chain-pairing” problem. As summarized in Table 2, various genetic alternations in the polypeptides, which introduce specific protein-protein interaction fits through shapes, charges or domain swaps between protein chains, have been proven successful in producing correctly assembled multispecific antibodies.
Table 1. Building blocks of multispecific biotherapeutics.
| Binding Modules | Binding Modules | Molecular Weight | Molecular Properties | References |
|---|---|---|---|---|
| Fragments of antibody (Fabs) |  |
~50 kDa | Medium half-life/less aggregated | [9][10] |
| Single-chain variable fragment (scFv) |
 |
~25 kDa | Short half-life/less aggregated | [9][10] |
| Diabodies (Db) |  |
~25 kDa | Short half-life/less aggregated | [9][10] |
| Single-domain antibodies (VHH from llama or camel, VNAR from shark) |  |
~15 kDa | Short half-life/less aggregated | [9][10] |
| Synthetic peptides |  |
~3 kDa | Extremely short half-life/less aggregated | [24][25][26][27] |
| TCR domains |  |
~50 kDa | Medium half-life/less aggregated | [28][29][30][31] |
| Enzyme domains |  |
~50 kDa | Medium half-life/less aggregated | [32] |
| Small scaffolds (Affibody, Fynomers, Monobodies, DARPins, Knottins, VLRs, Nanoantibodies) |  |
~5–12 kDa | Short half-life/less aggregated | [15][33][34][35][36][37][38][39] |
| Chemical molecules |  |
~3 kDa | Short half-life/aggregated | [6][40] |
| Oligonucleotides |  |
~10 kDa | Short half-life/less aggregated | [41][42] |
Table 2. Strategies and mutations to ensure cognate light and heavy chain assembly in bispecific antibodies with the numbering of the European Union amino acid sequence (All are in human IgG1 unless mentioned otherwise).
| Technology Name | Mutations in First Chain | Mutations in Second Chain | References |
|---|---|---|---|
| Knobs-into-holes (Genentech) |
HC1: S354C, T366W | HC2: Y349C, T366S, L368A, Y407V | [43][44] |
| Electrostatic steering (Amgen) | HC1:K409D, K392D | HC2: D399K, E356K | [45] |
| Electrostatic steering (Pfizer) | IgG1 HC1: D221E, P228E, L368E IgG2 HC1: C223E, P228E, L368E |
IgG1 HC2: D221R, P228R, K409R IgG2 HC2: C223R, E225R, P228R, K409R |
[46] |
| Electrostatic Steering (Merus) |
HC1: L351D, L368E | HC2: L351K, T366K | [47] |
| Fab-arm exchange (Genmab) | HC1: K409R | HC2: F405L | [48][49] |
| SEED (EMD Serono) |
HC1: IgG/IgA chimera |
HC2: IgG/IgA chimera |
[50] |
| LUZ-Y (Genentech) | HC1: cleavable leucine zipper | HC2: cleavable leucine zipper | [51] |
| HA-TF (Xencor) |
HC1: S364H, F405A | HC2: Y349T, T394F | [52] |
| EW-RVT (EW-RVTs-s) (Ajou University) |
HC1: K360E, K409W (Y349C) | HC2: Q347R, D399V, F405T (S354C) | [53][54] |
| ZW1 (VYAV-VLLW) (Zymeworks) |
HC1: T350V, L351Y, F405A, Y407V | HC2: T350V, T366L, K392L, T394W | [55] |
| DMA-RRVV (SYMV-GDQA) (UNC/Eli Lily) |
HC1:K360D, D399M, Y407A (Y349S, K370Y, T366M, K409V) |
HC2: E345R, Q347R, T366V, K409V (E356G, E357D, S364Q, Y407A) |
[56] |
| Protein A affinity (Regeneron) | HC1: H435R | None | [57] |
| Protein A and Protein G Avidity (Glenmark) |
HC1: IgG3Fc, N82aS | HC2: M428G/N434A/K213V | [58] |
| CrossMab (Roche) |
HC1:CL-VH | LC1:CH1-VL | [59] |
| Fab-Interface engineering (Eli Lily) |
HC1: Q39K, R62E, H172A, F174G LC1: D1R, Q38D, L135Y, S176W |
HC2: Q39Y LC2: Q38R |
[60] |
| Fab-Interface electrostatic steering (Amgen) |
HC1: Q39K, Q105K, S183D LC1: Q38D, A43D, S176K |
HC2: Q39D, Q105D, S183K LC2: Q38K, A43K, S176D |
[61] |
| Κλ-bodies (Novimmune SA) |
LC1:κ | LC2:λ | [62] |
| Common Light Chain (Genentech & Merck KGaA) |
Shared LC | Shared LC | [63][64] |
| Tetravalent IgG-like Charged CR3 mutant (Biomunex) | Mab1 CH1: T192E | Mab1 CL: N137K, S114A |
[65] |
| Tetravalent IgG-like Hydrophobicity-polarity swap MUT4 mutant (Biomunex) |
Mab1 CH1: L143Q, S188V | Mab1 CL: V133T, S176V |
[65] |
| Fabs-in-Tandem (FIT-Ig) (EpimAb) | Long chain: VLA-CL-VHB-CH1-CH2-CH3 | Short Chain A: VHA-CH1, Short Chain B:VLB-CL |
[66] |
| DuetMab (AstraZeneca) |
CH1: F126C | CL: S121C | [67][68] |
| BEAT (Glenmark) |
HC1-CH3: Residues from TCR α interface |
HC2-CH3: Residues from TCR β interface |
[69] |
| TCR CαCβ (Eli Lily) |
HC1-CH1: TCR Cα |
LC1:CL: TCR Cβ |
[30] |
| WuXiBody (WuXi Biologics) |
HC1-CH1: TCR Cβ |
LC1-CL: TCR Cα |
[31] |
Non-protein entities utilize small molecules and oligonucleotides to greatly broaden the mechanism of action and design versatilities of the multispecific biotherapeutics [6][70][40][71][41]. They enable specific binding to intracellular drug targets such as those involved in replication and division of cells and DNAs. These non-protein moieties are covalently linked to protein backbones (typically antibodies) that target specific surface antigens in cells and tissues. Upon the arrival to either the extracellular space or inside cells through endocytosis, the conjugated chemical molecules or oligonucleotides are released to exert therapeutic effects.
The current generation of multispecific innovative drugs have ridden a wave of promising clinical and regulatory successes. As of January 2021, thirteen multispecific molecules have been commercialized. Among them, there are three approved bispecific antibodies (catumaxomab (RemovabTM), blinatumomab (Blincyto®), emicizumab (Hemlibra®)), one approved antibody binding fragment fusion with immunotoxin (LumoxitiTM), and nine approved antibody-drug conjugates (gemtuzumab ozogamicin (Mylotarg®), brentuximab vedotin (Adcetris®), ado-trastuzumab emtansine (Kadcyla®), inotuzumab ozogamicin (Besponsa®), enfortumab vedotin (PadcevTM), fam-trastuzumab deruxtecan-nxki (Enhertu®), polatuzumab vedotin-piiq (PolivyTM), belantamab mafodotin-blmf (BlenrepTM), sacituzumab govitecan (TrodelvyTM)). These successes, along with nearly 200 multispecific biotherapeutic candidates in the clinical pipelines [3][4][5][6][10][70][40][71][41][72][15][73][74][75][76][77][78][79], indicate that multispecific biotherapeutics are promising molecules. However, discovering and developing them into an approved product is challenging and requires extensive efforts in engineering and development. The following sections summarize the recent advances of the major classes of multispecific biotherapeutics drugs (Figure 1) in designs and applications.
2.1. Immune-Cell Engagers
Directing immune cells to target tumor cells represents one major group of mechanism of action for the multispecific biotherapeutics. These so-called immune-cell engagers are the quintessential example of the obligate multispecific biotherapeutics which take advantage of the presence of different binding specificities within a single polypeptide molecule to achieve a new function. They are designed to form an artificial cytolytic synapse by bringing immune cells, such as cytotoxic T-cells and natural killer (NK) cells, into close proximity with tumor cells [4][5][11][74]. Subsequent release of granzymes and perforin from activated T or NK cells into tumor cells results in tumor cell death. In the meantime, activated NK cells can also evoke target cell caspase activation via TNF-related apoptosis-inducing ligand and Fas ligand. Versatile factors can be secreted by NK cells for modulating functions of other immune cells.
The formats for the immune-cell engagers are generally classified into IgG-like molecules and non-IgG-like molecules [4][5][9][10], each with advantages and disadvantages. IgG-like molecules retain the Fc component that may endow effector functions and FcRn-mediated recycling for longer serum half-lives, whereas large sizes of these molecules could hinder their tissue penetration. To generate multispecific IgG-like engagers that interact with both immune cells and tumor cells, a number of antibody formats have been reported. Quadroma relies on the fusion of two distinct hybridomas [80], through which catumaxomab is produced in a rat/mouse quadroma cell line. Engineering chain-pairing scaffolds with “Knobs-into-holes” [43][44] have been widely used [81][82][83], whereas electrostatic steering strategies [45][46] have been shown with successful applications [84][85]. Dual-variable domains Ig (DVD-Ig) [86][87] (formed by the fusion of the variable domains of two antibodies in tandem), IgG-scFv [9][10] (produced by fusing scFv or Fv to the termini of antibody chains), κλ bodies [62] (sharing same heavy chain with two different light chains (κ or λ) with highly selective affinity resins for purification), trivalent format [88], and various tetravalent formats [65][66][89] have been utilized for the design of new immune-cell engagers.
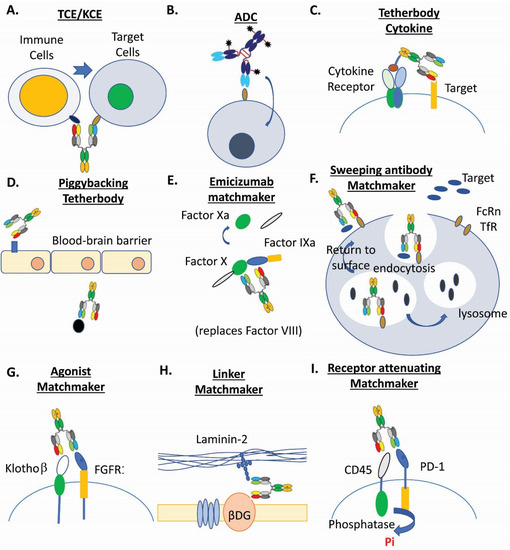
Figure 1. Examples of multispecific biotherapeutics drugs. (A). T-cell and NK-cell engagers (TCE/KCE) [4][5][11]. (B). Antibody-drug conjugates (ADC) [3][6][90][40][71][91]. (C). Tetherbody cytokine [92]. (D). Piggybacking tetherbody [93][94][95]. (E). Emicizumab matchmaker [96][97]. (F). Sweeping antibody matchmaker [3][98]. (G). Agonist matchmaker [99][100]. (H). Linker matchmaker [101]. (I). Receptor attenuating matchmaker [102].
Non-IgG-like molecules do not contain Fc, but rather have different antigen binding fragments. They can become dimers, trimers, or tetramers depending on linker length and fragment sequences. A number of non-IgG-like formats have been reported: scFv-based molecules (exemplified by blinatumomab with the fusion protein containing light-chain variable domain (VL) and heavy-chain variable domain (VH), or tandem scFvs), diabody format (single-chain diabody, tandem diabodies, exemplified by dual-affinity retargeting molecules (DART)), and nanobodies from llamas, camels, or shark. The smaller size of the non-IgG-like formats offers advantages like enhancing tissue penetration, ease of manufacturing, and accessing sterically hindered epitopes. Their drawbacks are short half-lives and poor target-site retention. Besides the improvement with constant administration in the case of blinatumomab, a number of strategies have been put in place for serum half-life extension [103]. Attachment of highly flexible and hydrophilic molecules such as polyethylene glycol, carbohydrates, N-(2-hydroxypropyl) methacrylamide, dextran, and amino acid polymer XTEN [104] can increase the hydrodynamic volume. Fusions with human serum albumin (HSA) or albumin-binding domain like scFv to HSA can increase half-life significantly [105][106]. Additionally, multimerization can increase sizes and binding valency of the molecules [10][107].
2.2. Antibody Drug Conjugates
Antibody drug conjugates (ADCs) (Table 3) are an important class of multispecific biotherapeutics that combine two or more binding activities into one entity [3][6][90][40][71][91]. They are composed of chemical compounds (cytotoxic drugs or nucleotide acids) covalently linked to an antibody component that binds to a specific target. The selective binding to the target brings the conjugated chemical components into specific cells or tissues where they can exert their therapeutic effects. This should significantly limit the undesirable side effects of cytotoxic drugs or oligonucleotides. ADCs are often considered as prodrugs that are supposed to be nontoxic in circulation. A key attribute for an ADC is the average number of payloads (conjugated drugs) per antibody or the so-called drug-to-antibody ratio (DAR). The DAR can affect an ADCs’ potency and toxicity as well as plasma stability, biophysical properties, and pharmacokinetics.
Table 3. Target antigens, antibody, conjugation methods, linkers, payloads, and average drug antibody ratio (DAR) of approved antibody-drug-conjugates.
| ADC Products | Target Antigen/Antibody | Conjugation Methods (Lys/Cys) Random/Site-Specific | Linker Payload | Average Drug Antibody Ratio |
|---|---|---|---|---|
| gemtuzumab ozogamicin (Pfizer) |
CD33/humanized IgG4κ | Lys/random | N-acetyl-γ calicheamicin 1,2 dimethyl hydrazine dichloride | ~1.5 |
| brentuximab vedotin (Seattle Genetics) | CD30/chimeric IgG1 | Interchain Cys/random | mc-vc-PABC-MMAE | ~4 |
| ado-trastuzumab emtansine (Roche) |
HER2/humanized IgG1 | Lys/random | SMCC-DM1 | ~3.5 |
| inotuzumab ozogamicin (Pfizer) |
CD22/humanized IgG4 | Lys/random | N-acetyl-γ calicheamicin 1,2 dimethyl hydrazine dichloride | ~5–7 |
| enfortumab vedotin (Astellas) | Nectin-4/human IgG1κ | Interchain Cys/random | mc-vc-PABC-MMAE | ~4 |
| fam-trastuzumab deruxtecan-nxki (Daiichi Sankyo) | HER2/humanized IgG1κ | Interchain Cys/site-specific | mc-GGFG-DX-8951 derivative | ~7.7 |
| polatuzumab vedotin-piiq (Roche) |
CD79b/humanized IgG1κ | Interchain Cys/random | mc-vc-PABC-MMAE | ~4 |
| belantamab mafodotin-blmf (GlaxoSmithKline) |
BCMA/Afucosylated humanized IgG1 | Interchain Cys/random | mc-MMAF | ~4 |
| sacituzumab govitecan (Immunomedics) |
TROP2/humanized IgG1κ | Interchain Cys/random | Cl2A-SN38 | ~7.6 |
Similar to that of immune-cell engagers, the successful development of an ADC depends on the selection of an appropriate target antigen for the antibody component. Besides high tumor-association to minimize on-target toxicity of ADCs, the antigens engaged by ADCs need to be endocytosed upon antibody binding. High copy numbers of antigens’ surface expression in tumor cells (>105/cell, [108]) is crucial for the efficacy of ADCs because as more surface antigens bind to the ADC, then more chemical drugs can be delivered into the tumor cells [6][40]. Since most TAAs also express in normal tissues at lower levels, bispecific ADCs targeting two TAAs should increase tumor cell specificity. Such molecules have been recently shown to increase ADCs’ internalization and reduces tumor resistance in preclinical models [109].
For designing an effective ADC, selection criteria for cytotoxic payloads should include strong cell toxicity, preservation of potency after conjugation release, acceptable aqueous solubility, stability in aqueous formulation and physiological conditions, as well as synthetic feasibility under good manufacturing practice [6][90][40][71][91]. Most cytotoxic drugs are hydrophobic and tend to induce antibody aggregation which cause fast clearance and acute host responses [40][110]. The current major categories for cytotoxic payloads are microtubule inhibitors and DNA-damaging agents. Antimitotic agents cause cell death by interfering with the ability of mitotic spindles to segregate chromosomes and alter cellular cytoskeletal architecture [108][111]. The two most widely used antimitotic agents for ADCs are based on auristatins and maytansinoids [108][111]. For DNA-damaging agents, they are DNA binding agents that attach to the double-helix and promote DNA strand alkylation, scission or crosslinking [108][111]. The representative examples for this agent class are calicheamicins, duocarmycins, camptothecins, anthracyclines, and pyrrolobenzodiazepines. Other mechanisms of action for ADCs’ cytotoxic payloads include Topoisomerase I inhibitor-based payload, RNA splicing inhibition using natural product-derived thailanstatin, and RNA polymerase I inhibition using α-amanitin derivatives [71]. Besides small molecule payloads, synthetic oligonucleotide therapeutics (antisense oligonucleotides and siRNA) [41][42][112] have been conjugated into the antibody backbone showing efficacy in vivo and in vitro [113][114][115]. These payloads can target the RNAs encoding those intracellular proteins that are inaccessible and un-druggable by antibodies alone.
Linkers that connect payloads to antibodies are another critical feature of ADCs [6][90][40][71][91]. They must be stable in circulation and able to release the drugs in active form in the targeted location. The intrinsic features of the linkers and the payloads determine the physicochemical properties of ADC metabolites in nearby cells. As a result, the linkers induce a “bystander effect” to kill neighboring cells [6][90][40][71][91]. They can be classified into non-cleavable (like the one in trastuzumab emtansine) and cleavable (like the one in brentuximab vedotin). Non-cleavable linkers remain attached to the drugs after proteolytic degradation of the antibody in lysosome, which with linkers and an amino acid or short peptide are released into the cytosol to exert their activities [116]. The most common example of a non-cleavable linker is the thioether linker. For cleavable linkers, they exploit the differences between bloodstream and cytoplasm of targeted tissues. They can be cleaved by certain specific proteases found in lysosomes, or by low pH (acidic environment), or by a reducing environment (high glutathione concentrations in cytoplasm) to release free drugs [117]. Peptide linkers, hydrazone, and disulfide are a typical type of cleavable linkers.
Conjugation strategy is another important aspect for ADCs. Linker-payload moieties are typically conjugated to antibodies via lysine or cysteine residues. Ideally, the conjugation process should maintain the integrity of antibodies and the activity of drugs, as well as produce homogeneous batches that remain stable during the storage and lyophilization process [40]. Because the IgG scaffold has over 80 lysine residues, about 20 of which are highly solvent-accessible [118], the conjugation process results in highly heterogenous mixtures. For cysteine conjugation, four interchain disulfides in one IgG1 also lead to a heterogeneous ADC [119]. More homogeneous ADCs can be produced through site-specific conjugation via genetic engineering [6][90][40][71][91][120][121]. Engineered cysteine substitution at light chain and heavy chain positions provides reactive thiols and allows site-specific drug conjugation [122][123][124][125]. Unnatural amino acids capable of bio-orthogonal chemical conjugation can be introduced to antibodies through amber codon suppression [126]. Transglutaminase couple glutamine residues side-chain with linker-payloads through amide bonds [127][128]. Sortase catalyzed the ligation of the LPXTG tag at the C-terminus of a polypeptide with the N-terminal oligoglycine [129]. Glycotransferases can be engineered to transfer modified monosaccharides that enable bio-orthogonal chemical conjugation to a linker-payload [130]. Formylglycine-generating enzyme can covert an engineered cysteine residue in a specific peptide sequences such as CXPXR to produce an aldehyde tag for linker conjugation [131]. Most recently, engineered methionine residues have been utilized for efficient site-specific drug conjugation [132][133], and ADP-ribosyl cyclases enable a facile approach for generating site-specific ADXs CD38 fusion proteins [32].
2.3. Tetherbodies
Tetherbodies are a class of multispecific biotherapeutics drugs that utilize one binding interface to engage a docking protein for action enrichment to a particular location, with another binding interface to engage an effector protein. Regarding the mechanism of action, tetherbodies are similar to ADC, but its modalities are all protein-based and produced by recombinant DNAs [3].
The typical examples for tetherbodies are antibody-immunotoxin fusion (LumoxitiTM) or antibody-cytokine fusion [92]. Immuotoxin or Cytokines (such as IL-2) have adverse effects when administered systemically. When fused to an antibody that binds to a TAA, immunotoxin or cytokines like IL-2, tetherbodies can be enriched and interact with receptors at those tumor sites. This interaction can greatly limit the toxicities of the resulting tetherbodies. Another example for tetherbodies is the “piggyback” or “hijacking” approach [3][4]. Because blood-brain barrier (BBB) restricts access of large molecules to the brain parenchyma, multispecific biotherapeutics with one antibody arm against plasma membrane receptors, e.g., transferrin receptor (TfR) that transcytose through brain endothelial cells, have been explored for efficient brain entry [93][94][95][134][135]. The efficacies of these tetherbodies have been demonstrated in animal models [94][135], yet clinical successes remain to be seen. Identifying novel receptors that are brain endothelial cell-specific represents a new opportunity. A similar piggyback mechanism for tetherbodies has been used for the indication of infectious diseases. By targeting persistent factor Psl and the needle tip protein PcrV of Pseudomonas aeruginosa [136], the resulting bsAb can increase internalization and localization in acidic vacuoles for neutrophil killing. In addition, by utilizing one arm targeting an extracellular exposed epitope in the filovirus glycoprotein (GP) of Ebola virus to gain access to endosome during viral uptake [137], a “Trojan horse” bispecific antibody used the other arm against a cryptic receptor binding site of GP or the corresponding Niemann-Pick C1 intracellular receptor to block virus entry to cytoplasm.
One novel subclass of tetherbody-like multispecific biotherapeutics drugs is the condition-activated tetherbody that can enrich the active drugs in a particular location through the design of conditional activation. The conditional activation mechanisms take advantage of special conditions in tumor microenvironment such as increased levels of specific proteases [138], ATP [139][140], and others [72]. Because the presence of tumor antigen in healthy tissues can cause on-target toxicity by ADCs or bsTCEs, ProbodiesTM (CytomX) with a protease-cleavable peptide linker and a masking peptide can presumably remain inert until proteolytically activated locally in disease tissues [138]. A novel class of ADC called Probody-drug conjugates has shown the potential to protect normal tissues expressing target antigens [141]. The probody for EGFRxCD3 bsTCE has reported a 60-fold increase in dose tolerance compared to the unmasked construct [72][76]. A similar conditional activated TCE called ProTIA (protease-triggered immune activator) has been developed by Amunix and its AMX-168 is an anti-EpCAM BiTE-TCE fused to a masking XTEN polymer by a protease-cleavable linker [76].
2.4. Biologic Matchmaker Drugs
Biologic matchmaker drugs are a relatively new class of multispecific biotherapeutics that utilize one binding interface to engage an action partner, with another binding interface to engage a target [3]. They bring the drug targets in close proximity to an action partner for efficient biological activation or inactivation.
The best known example of a biologic matchmaker drug is emicizumab that replaces the scaffold function of factor VIII for generating activated FX [96][97]. Through hetero-binding arms, this bispecific antibody brings the substrate FX to the protease enzyme FIXa. A similar biologic matchmaker is a molecular linker that connects laminin-211 and the dystroglycan beta-subunit [101]. A novel bispecific antibody targeting both laminin and dystroglycan proteins has been developed to ameliorate sarcolemmal fragility for improving muscle function [101].
Biomimetics for signaling molecules are another type of biologic matchmaker. Bispecific agonistic antibodies that engage with FGFR1 with one arm and Klothoβ with another arm [99][100] have been developed to mimic fibroblast growth factor 21 for its signaling action. Most recently, bispecific biologic matchmakers have been constructed to recruit promiscuous cell-surface phosphatase CD45 to attenuate signaling of kinase-activated cell surface receptors such as PD-1 and SIRPα through dephosphorylation [102].
There are also other novel formats for biologic matchmaker drugs in early stage preclinical research. One example is the multispecific biotherapeutics that utilized the mechanism similar to heterobifunctional proteolysis-targeting chimeric molecules (PROTACs) [142][143][144]. Through the PROTACs’ first ligand to the target and the second ligand to ubiquitin ligase, PROTACs induce ubiquitylation of the target followed by proteolytic degradation in the proteasome [3]. A similar PROTACs-like multispecific biotherapeutics has been produced by Kanner et al. [145]. This PROTACs-like matchmaker consisted of a llama single-chain antibody fused to the catalytic domain of E3 ligase which can increase targeted ubiquitination of ion channels and consequentially diminish the surface expression of these membrane proteins. Interestingly, an opposite application has also been recently reported by the same research team. A multispecific deubiquitinase, in which a deubiquitinase was fused to a nanobody, deubiquitinated trafficking-deficient ion channels and consequentially rescued their functional surface expression [146]. Similarly, a multispecific matchmaker, named sweeping antibody, utilizes a lysosomal degradation system without taking advantage of proteasome activity [3]. This multispecific matchmaker binds to either FcRn or a surface antigen (i.e., TfR) in one arm at both neutral and acidic pH, with another arm interacting with the target only at neutral pH [3][98]. When the multispecific is endocytosed to endosome, the target is then released and proceeds to lysosome for degradation.
2.5. Small-Scaffold Multispecific Modalities
As an alternative to antibodies, various scaffold proteins have been developed and utilized for multispecific targeting [15][16]. They are Fynomers, Monobodies, Affibodies, DARPins, Knottins, Anticalins, synthetic peptides, CH2-based nanoantibodies, and others. These small-scaffolds have the advantages of small size, good stability, good tissue penetration, and reasonably high binding affinity, serving as robust building blocks for multispecific biotherapeutics.
Fynomers are among those best-characterized small-scaffolds that can be engineered to bind to targets of choice with antibody-like affinity and specificity [147]. They are small stable globular proteins (~7 kDa) derived from the SH3 domain of human Fyn kinase without disulfide bonds [147]. A bispecific antibody (FynomAb) was generated by fusing a HER2-specific Fynomer to either the N or C-terminus of a heavy or light chain of pertuzumab [38], showing a superior anti-tumor activity. COVA322 is a clinical-stage bispecific TNF/IL17-A inhibitor with IL-17A-specfic Fynomer fused to adalimumab [148], displaying good efficacy for inflammatory diseases.
Monobodies’ scaffold folding is similar to immunoglobulin domains but does not rely on the formation of an intradomain disulfide bond [149][150]. They are small scaffolds (~10 kDa) derived from fibronectin type II domain (FN3) [149][151]. Centyrins are structurally similar to Monobodies, but from the FN3 domain of Tenascin C [152]. A single loop and the face of a β-sheet form their diverse binding surfaces [33][150][153]. A bispecific Monobody-ubiquitin ligase fusion was generated for target degradation [154]. Two anti-EGFR Centyrin cytotoxic conjugates showed good IC50 values of around 0.2 nM [155].
Affibodies can be functionally produced both by peptide synthesis and by recombinant bacterial expression [156]. They are small scaffolds (~7 kDa) derived from a 58-amino-acid alpha-helical Z-domain of Staphylococcus protein-A [34][157][158]. They can display high affinity to a variety of targets such as HER2, selected from combinatorial libraries [156]. Anti-HER2 cytotoxic Affibody conjugate was generated and showed inhibition activity on tumor growth [159]. Recently, a trispecific affibody fusion protein consisting of anti-amyloid beta, a scFv against TfR and an albumin binding domain, was shown to increase uptake into the cerebrospinal fluid 24 h after injection [160].
Designed ankyrin repeat proteins (DARPins) are small stable scaffolds (~15–18 kDa) derived from Akyrin repeats which utilize their repetitive structural units to form an extended binding surface [161]. Anti-EpCAM DARPin toxin fusion [162] and anti-HER2 toxin fusion [163] have shown strong anti-tumor activity in vivo. Recently, a novel bispecific anti-EGFR DARPin fused with the inhibitory prodomain of ADAM17 was shown to decrease cell proliferation and migration of EGFR-dependent cancer cells [164]. MP0250, a trispecific DARPin in a Phase I/II study for advanced solid tumors, can bind to VEGF-A and hepatocyte growth factor as well as human serum albumin [73][165]. Anti-EpCAM DARPin fused to arginine-rich Low Molecular Weight Protamine protein was used for efficient siRNA delivery [166].
Knottins (~3–6 kDa) are cysteine knots with 30–50 amino acid residues exhibiting excellent stability [37]. Integrin-targeting knottin peptide-drug-conjugates showed potent inhibition on tumor cell proliferation [167]. Variable lymphocyte receptors (VLRs) are utilized by jawless vertebrates for adaptive immune recognition [36], and contain leucine-rich repeats found in mammalian innate immune receptors. They have been developed as binding scaffolds to therapeutic targets like IL6 [168]. Anticalins are derived from lipocalins and exhibit a structurally conserved αβ-barrel with an attached α-helix supporting hypervariable loops to bind protein molecules [169]. PRS-343, an agonistic anticalin targeting costimulatory receptor 4–1BB with an antibody against human HERs, are currently in clinical-stage development [170].
Human Ig constant CH2 domains (CH2 of IgG, IgA and IgD, and CH3 of IgE and IgM) have been proposed and utilized as a novel small-binding scaffold [17]. They have been coined nanoantibodies as they can be diversified by random mutagenesis and contain binding sites with stability and effector functions. Bispecific nanoantibodies have been engineered to interact with FcRn and HIV-1 neutralizing epitope [39].
Specific peptides can also be used for the fusion to an IgG to generate multispecific molecules either through genetic fusion [24][25] or chemical conjugation with a catalytic antibody called CovX-Bodies [26][171][172]. The small size of peptides provide advantages of epitope access and design versatility [27]. Zybody technology enables up to five target-specificities [25], but their degradation susceptibility requires special chemical modifications [24][172]. Peptide-conjugates utilize peptides such as those derived from natural receptors [173] or bicyclic peptides have shown better tumor penetration [71]. Peptide-antibody fusions have been recently generated against Middle East Respiratory Syndrome Coronavirus [174].
References
- Hodgson, J. Refreshing the biologic pipeline 2020. Nat. Biotechnol. 2021, 39, 135–143.
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018, 17, 197–223.
- Deshaies, R.J. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature 2020, 580, 329–338.
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608.
- Goebeler, M.E.; Bargou, R.C. T cell-engaging therapies—BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434.
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337.
- Nisonoff, A.; Rivers, M.M. Recombination of a mixture of univalent antibody fragments of different specificity. Arch. Biochem. Biophys. 1961, 93, 460–462.
- Aalberse, R.C.; van der Gaag, R.; van Leeuwen, J. Serologic aspects of IgG4 antibodies. I. Prolonged immunization results in an IgG4-restricted response. J. Immunol. 1983, 130, 722–726.
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67, 95–106.
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212.
- Thakur, A.; Huang, M.; Lum, L.G. Bispecific antibody based therapeutics: Strengths and challenges. Blood Rev. 2018, 32, 339–347.
- Staerz, U.D.; Kanagawa, O.; Bevan, M.J. Hybrid antibodies can target sites for attack by T cells. Nature 1985, 314, 628–631.
- Perez, P.; Hoffman, R.W.; Shaw, S.; Bluestone, J.A.; Segal, D.M. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature 1985, 316, 354–356.
- Nagorsen, D.; Kufer, P.; Baeuerle, P.A.; Bargou, R. Blinatumomab: A historical perspective. Pharmacol. Ther. 2012, 136, 334–342.
- Jost, C.; Pluckthun, A. Engineered proteins with desired specificity: DARPins, other alternative scaffolds and bispecific IgGs. Curr. Opin. Struct. Biol. 2014, 27, 102–112.
- Gilbreth, R.N.; Koide, S. Structural insights for engineering binding proteins based on non-antibody scaffolds. Curr. Opin. Struct. Biol. 2012, 22, 413–420.
- Dimitrov, D.S. Engineered CH2 domains (nanoantibodies). MAbs 2009, 1, 26–28.
- Raghavan, M.; Bonagura, V.R.; Morrison, S.L.; Bjorkman, P.J. Analysis of the pH dependence of the neonatal Fc receptor/immunoglobulin G interaction using antibody and receptor variants. Biochemistry 1995, 34, 14649–14657.
- Raghavan, M.; Bjorkman, P.J. Fc receptors and their interactions with immunoglobulins. Annu. Rev. Cell Dev. Biol. 1996, 12, 181–220.
- Krah, S.; Kolmar, H.; Becker, S.; Zielonka, S. Engineering IgG-Like Bispecific Antibodies-An Overview. Antibodies 2018, 7, 28.
- Santich, B.H.; Park, J.A.; Tran, H.; Guo, H.F.; Huse, M.; Cheung, N.K. Interdomain spacing and spatial configuration drive the potency of IgG-[L]-scFv T cell bispecific antibodies. Sci. Transl. Med. 2020, 12, eaax1315.
- Krah, S.; Sellmann, C.; Rhiel, L.; Schröter, C.; Dickgiesser, S.; Beck, J.; Zielonka, S.; Toleikis, L.; Hock, B.; Kolmar, H.; et al. Engineering bispecific antibodies with defined chain pairing. New Biotechnol. 2017, 39, 167–173.
- Sun, W.; Khare, P.; Wang, X.; Challa, D.K.; Greenberg, B.M.; Ober, R.J.; Ward, E.S. Selective Depletion of Antigen-Specific Antibodies for the Treatment of Demyelinating Disease. Mol. Ther. 2021, 29, 1312–1323.
- Zhong, X.; Kieras, E.; Sousa, E.; D’Antona, A.; Baber, J.C.; He, T.; Desharnais, J.; Wood, L.; Luxenberg, D.; Stahl, M.; et al. Pyroglutamate and O-Linked Glycan Determine Functional Production of Anti-IL17A and Anti-IL22 Peptide-Antibody Bispecific Genetic Fusions. J. Biol. Chem. 2013, 288, 1409–1419.
- Kanakaraj, P.; Puffer, B.A.; Yao, X.T.; Kankanala, S.; Boyd, E.; Shah, R.R.; Wang, G.; Patel, D.; Krishnamurthy, R.; Kaithamana, S.; et al. Simultaneous targeting of TNF and Ang2 with a novel bispecific antibody enhances efficacy in an in vivo model of arthritis. MAbs 2012, 4, 600–613.
- Cao, Y.; Suresh, M.R. Bispecific antibodies as novel bioconjugates. Bioconjug. Chem. 1998, 9, 635–644.
- Trier, N.; Hansen, P.; Houen, G. Peptides, Antibodies, Peptide Antibodies and More. Int. J. Mol. Sci. 2019, 20, 6289.
- Lowe, K.L.; Cole, D.; Kenefeck, R.; OKelly, I.; Lepore, M.; Jakobsen, B.K. Novel TCR-based biologics: Mobilising T cells to warm ‘cold’ tumours. Cancer Treat. Rev. 2019, 77, 35–43.
- Oates, J.; Hassan, N.J.; Jakobsen, B.K. ImmTACs for targeted cancer therapy: Why, what, how, and which. Mol. Immunol. 2015, 67, 67–74.
- Wu, X.; Sereno, A.J.; Huang, F.; Zhang, K.; Batt, M.; Fitchett, J.R.; He, D.; Rick, H.L.; Conner, E.M.; Demarest, S.J. Protein design of IgG/TCR chimeras for the co-expression of Fab-like moieties within bispecific antibodies. MAbs 2015, 7, 364–376.
- Guo, G.; Han, J.; Wang, Y.; Li, Y. A potential downstream platform approach for WuXiBody-based IgG-like bispecific antibodies. Protein Expr. Purif. 2020, 173, 105647.
- Dai, Z.; Zhang, X.N.; Nasertorabi, F.; Cheng, Q.; Li, J.; Katz, B.B.; Smbatyan, G.; Pei, H.; Louie, S.G.; Lenz, H.J.; et al. Synthesis of site-specific antibody-drug conjugates by ADP-ribosyl cyclases. Sci. Adv. 2020, 6, eaba6752.
- Sha, F.; Salzman, G.; Gupta, A.; Koide, S. Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 2017, 26, 910–924.
- Ståhl, S.; Gräslund, T.; Karlström, A.E.; Frejd, F.Y.; Nygren, P.Å.; Löfblom, J. Affibody Molecules in Biotechnological and Medical Applications. Trends Biotechnol. 2017, 35, 691–712.
- Lipovšek, D.; Carvajal, I.; Allentoff, A.J.; Barros, A., Jr.; Brailsford, J.; Cong, Q.; Cotter, P.; Gangwar, S.; Hollander, C.; Lafont, V.; et al. Adnectin-drug conjugates for Glypican-3-specific delivery of a cytotoxic payload to tumors. Protein Eng. Des. Sel. 2018, 31, 159–171.
- Alder, M.N.; Rogozin, I.B.; Iyer, L.M.; Glazko, G.V.; Cooper, M.D.; Pancer, Z. Diversity and function of adaptive immune receptors in a jawless vertebrate. Science 2005, 310, 1970–1973.
- Kintzing, J.R.; Cochran, J.R. Engineered knottin peptides as diagnostics, therapeutics, and drug delivery vehicles. Curr. Opin. Chem. Biol. 2016, 34, 143–150.
- Brack, S.; Attinger-Toller, I.; Schade, B.; Mourlane, F.; Klupsch, K.; Woods, R.; Hachemi, H.; von der Bey, U.; Koenig-Friedrich, S.; Bertschinger, J.; et al. A bispecific HER2-targeting FynomAb with superior antitumor activity and novel mode of action. Mol. Cancer Ther. 2014, 13, 2030–2039.
- Gong, R.; Wang, Y.; Ying, T.; Dimitrov, D.S. Bispecific engineered antibody domains (nanoantibodies) that interact noncompetitively with an HIV-1 neutralizing epitope and FcRn. PLoS ONE 2012, 7, e42288.
- Abdollahpour-Alitappeh, M.; Lotfinia, M.; Gharibi, T.; Mardaneh, J.; Farhadihosseinabadi, B.; Larki, P.; Faghfourian, B.; Sepehr, K.S.; Abbaszadeh-Goudarzi, K.; Abbaszadeh-Goudarzi, G.; et al. Antibody-drug conjugates (ADCs) for cancer therapy: Strategies, challenges, and successes. J. Cell. Physiol. 2019, 234, 5628–5642.
- Leung, D.; Wurst, J.M.; Liu, T.; Martinez, R.M.; Datta-Mannan, A.; Feng, Y. Antibody Conjugates-Recent Advances and Future Innovations. Antibodies 2020, 9, 2.
- Benizri, S.; Gissot, A.; Martin, A.; Vialet, B.; Grinstaff, M.W.; Barthélémy, P. Bioconjugated Oligonucleotides: Recent Developments and Therapeutic Applications. Bioconjug. Chem. 2019, 30, 366–383.
- Atwell, S.; Ridgway, J.B.; Wells, J.A.; Carter, P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J. Mol. Biol. 1997, 270, 26–35.
- Ridgway, J.B.; Presta, L.G.; Carter, P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621.
- Gunasekaran, K.; Pentony, M.; Shen, M.; Garrett, L.; Forte, C.; Woodward, A.; Ng, S.B.; Born, T.; Retter, M.; Manchulenko, K.; et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: Applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 2010, 285, 19637–19646.
- Strop, P.; Ho, W.H.; Boustany, L.M.; Abdiche, Y.N.; Lindquist, K.C.; Farias, S.E.; Rickert, M.; Appah, C.T.; Pascua, E.; Radcliffe, T.; et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J. Mol. Biol. 2012, 420, 204–219.
- De Nardis, C.; Hendriks, L.J.; Poirier, E.; Arvinte, T.; Gros, P.; Bakker, A.B.; de Kruif, J. A new approach for generating bispecific antibodies based on a common light chain format and the stable architecture of human immunoglobulin G1. J. Biol. Chem. 2017, 292, 14706–14717.
- Labrijn, A.F.; Rispens, T.; Meesters, J.; Rose, R.J.; den Bleker, T.H.; Loverix, S.; van den Bremer, E.T.; Neijssen, J.; Vink, T.; Lasters, I.; et al. Species-specific determinants in the IgG CH3 domain enable Fab-arm exchange by affecting the noncovalent CH3-CH3 interaction strength. J. Immunol. 2011, 187, 3238–3246.
- Labrijn, A.F.; Meesters, J.I.; de Goeij, B.E.; van den Bremer, E.T.; Neijssen, J.; van Kampen, M.D.; Strumane, K.; Verploegen, S.; Kundu, A.; Gramer, M.J.; et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc. Natl. Acad. Sci. USA 2013, 110, 5145–5150.
- Davis, J.H.; Aperlo, C.; Li, Y.; Kurosawa, E.; Lan, Y.; Lo, K.M.; Huston, J.S. SEEDbodies: Fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng. Des. Sel. 2010, 23, 195–202.
- Wranik, B.J.; Christensen, E.L.; Schaefer, G.; Jackman, J.K.; Vendel, A.C.; Eaton, D. LUZ-Y, a novel platform for the mammalian cell production of full-length IgG-bispecific antibodies. J. Biol. Chem. 2012, 287, 43331–43339.
- Moore, G.L.; Bautista, C.; Pong, E.; Nguyen, D.H.; Jacinto, J.; Eivazi, A.; Muchhal, U.S.; Karki, S.; Chu, S.Y.; Lazar, G.A. A novel bispecific antibody format enables simultaneous bivalent and monovalent co-engagement of distinct target antigens. MAbs 2011, 3, 546–557.
- Choi, H.J.; Kim, Y.J.; Lee, S.; Kim, Y.S. A heterodimeric Fc-based bispecific antibody simultaneously targeting VEGFR-2 and Met exhibits potent antitumor activity. Mol. Cancer Ther. 2013, 12, 2748–2759.
- Choi, H.J.; Seok, S.H.; Kim, Y.J.; Seo, M.D.; Kim, Y.S. Crystal structures of immunoglobulin Fc heterodimers reveal the molecular basis for heterodimer formation. Mol. Immunol. 2015, 65, 377–383.
- Von Kreudenstein, T.S.; Escobar-Carbrera, E.; Lario, P.I.; D’Angelo, I.; Brault, K.; Kelly, J.F.; Durocher, Y.; Baardsnes, J.; Woods, R.J.; Xie, M.H.; et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: Quality by molecular design. MAbs 2013, 5, 646–654.
- Leaver-Fay, A.; Froning, K.J.; Atwell, S.; Aldaz, H.; Pustilnik, A.; Lu, F.; Huang, F.; Yuan, R.; Hassanali, S.; Chamberlain, A.K.; et al. Computationally Designed Bispecific Antibodies using Negative State Repertoires. Structure 2016, 24, 641–651.
- Smith, E.J.; Olson, K.; Haber, L.J.; Varghese, B.; Duramad, P.; Tustian, A.D.; Oyejide, A.; Kirshner, J.R.; Canova, L.; Menon, J.; et al. A novel, native-format bispecific antibody triggering T-cell killing of B-cells is robustly active in mouse tumor models and cynomolgus monkeys. Sci. Rep. 2015, 5, 17943.
- Ollier, R.; Wassmann, P.; Monney, T.; Ries Fecourt, C.; Gn, S.; CA, V.; Ayoub, D.; Stutz, C.; Gudi, G.S.; Blein, S. Single-step Protein A and Protein G avidity purification methods to support bispecific antibody discovery and development. MAbs 2019, 11, 1464–1478.
- Schaefer, W.; Regula, J.T.; Bähner, M.; Schanzer, J.; Croasdale, R.; Dürr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192.
- Lewis, S.M.; Wu, X.; Pustilnik, A.; Sereno, A.; Huang, F.; Rick, H.L.; Guntas, G.; Leaver-Fay, A.; Smith, E.M.; Ho, C.; et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat. Biotechnol. 2014, 32, 191–198.
- Liu, Z.; Leng, E.C.; Gunasekaran, K.; Pentony, M.; Shen, M.; Howard, M.; Stoops, J.; Manchulenko, K.; Razinkov, V.; Liu, H.; et al. A novel antibody engineering strategy for making monovalent bispecific heterodimeric IgG antibodies by electrostatic steering mechanism. J. Biol. Chem. 2015, 290, 7535–7562.
- Fischer, N.; Elson, G.; Magistrelli, G.; Dheilly, E.; Fouque, N.; Laurendon, A.; Gueneau, F.; Ravn, U.; Depoisier, J.F.; Moine, V.; et al. Exploiting light chains for the scalable generation and platform purification of native human bispecific IgG. Nat. Commun. 2015, 6, 6113.
- Jackman, J.; Chen, Y.; Huang, A.; Moffat, B.; Scheer, J.M.; Leong, S.R.; Lee, W.P.; Zhang, J.; Sharma, N.; Lu, Y.; et al. Development of a two-part strategy to identify a therapeutic human bispecific antibody that inhibits IgE receptor signaling. J. Biol. Chem. 2010, 285, 20850–20859.
- Krah, S.; Schröter, C.; Eller, C.; Rhiel, L.; Rasche, N.; Beck, J.; Sellmann, C.; Günther, R.; Toleikis, L.; Hock, B.; et al. Generation of human bispecific common light chain antibodies by combining animal immunization and yeast display. Protein Eng. Des. Sel. 2017, 30, 291–301.
- Golay, J.; Choblet, S.; Iwaszkiewicz, J.; Cérutti, P.; Ozil, A.; Loisel, S.; Pugnière, M.; Ubiali, G.; Zoete, V.; Michielin, O.; et al. Design and Validation of a Novel Generic Platform for the Production of Tetravalent IgG1-like Bispecific Antibodies. J. Immunol. 2016, 196, 3199–3211.
- Gong, S.; Ren, F.; Wu, D.; Wu, X.; Wu, C. Fabs-in-tandem immunoglobulin is a novel and versatile bispecific design for engaging multiple therapeutic targets. MAbs 2017, 9, 1118–1128.
- Mazor, Y.; Oganesyan, V.; Yang, C.; Hansen, A.; Wang, J.; Liu, H.; Sachsenmeier, K.; Carlson, M.; Gadre, D.V.; Borrok, M.J.; et al. Improving target cell specificity using a novel monovalent bispecific IgG design. MAbs 2015, 7, 377–389.
- Mazor, Y.; Hansen, A.; Yang, C.; Chowdhury, P.S.; Wang, J.; Stephens, G.; Wu, H.; Dall’Acqua, W.F. Insights into the molecular basis of a bispecific antibody’s target selectivity. MAbs 2015, 7, 461–469.
- Moretti, P.; Skegro, D.; Ollier, R.; Wassmann, P.; Aebischer, C.; Laurent, T.; Schmid-Printz, M.; Giovannini, R.; Blein, S.; Bertschinger, M. BEAT the bispecific challenge: A novel and efficient platform for the expression of bispecific IgGs. BMC Proc. 2013, 7 (Suppl. 6), O9.
- Shim, H. Bispecific Antibodies and Antibody-Drug Conjugates for Cancer Therapy: Technological Considerations. Biomolecules 2020, 10, 360.
- Gauzy-Lazo, L.; Sassoon, I.; Brun, M.P. Advances in Antibody-Drug Conjugate Design: Current Clinical Landscape and Future Innovations. SLAS Discov. 2020, 25, 843–868.
- Voynov, V.; Adam, P.J.; Nixon, A.E.; Scheer, J.M. Discovery Strategies to Maximize the Clinical Potential of T-Cell Engaging Antibodies for the Treatment of Solid Tumors. Antibodies 2020, 9, 65.
- Caputi, A.P.; Navarra, P. Beyond antibodies: Ankyrins and DARPins. From basic research to drug approval. Curr. Opin. Pharmacol. 2020, 51, 93–101.
- Strohl, W.R.; Naso, M. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies 2019, 8, 41.
- Clynes, R.A.; Desjarlais, J.R. Redirected T Cell Cytotoxicity in Cancer Therapy. Annu. Rev. Med. 2019, 70, 437–450.
- Ellerman, D. Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods 2019, 154, 102–117.
- Wu, Z.; Cheung, N.V. T cell engaging bispecific antibody (T-BsAb): From technology to therapeutics. Pharmacol. Ther. 2018, 182, 161–175.
- Middelburg, J.; Kemper, K.; Engelberts, P.; Labrijn, A.F.; Schuurman, J.; van Hall, T. Overcoming Challenges for CD3-Bispecific Antibody Therapy in Solid Tumors. Cancers 2021, 13, 287.
- Nie, S.; Wang, Z.; Moscoso-Castro, M.; D’Souza, P.; Lei, C.; Xu, J.; Gu, J. Biology drives the discovery of bispecific antibodies as innovative therapeutics. Antib. Ther. 2020, 3, 18–62.
- Seimetz, D.; Lindhofer, H.; Bokemeyer, C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat. Rev. 2010, 36, 458–467.
- Root, A.R.; Cao, W.; Li, B.; LaPan, P.; Meade, C.; Sanford, J.; Jin, M.; O’Sullivan, C.; Cummins, E.; Lambert, M.; et al. Development of PF-06671008, a Highly Potent Anti-P-cadherin/Anti-CD3 Bispecific DART Molecule with Extended Half-Life for the Treatment of Cancer. Antibodies 2016, 5, 6.
- Mathur, D.; Root, A.R.; Bugaj-Gaweda, B.; Bisulco, S.; Tan, X.; Fang, W.; Kearney, J.C.; Lucas, J.; Guffroy, M.; Golas, J.; et al. A Novel GUCY2C-CD3 T-Cell Engaging Bispecific Construct (PF-07062119) for the Treatment of Gastrointestinal Cancers. Clin. Cancer Res. 2020, 26, 2188–2202.
- Root, A.R.; Guntas, G.; Katragadda, M.; Apgar, J.R.; Narula, J.; Chang, C.S.; Hanscom, S.; McKenna, M.; Wade, J.; Meade, C.; et al. Discovery and optimization of a novel anti-GUCY2c x CD3 bispecific antibody for the treatment of solid tumors. MAbs 2021, 13, 1850395.
- Zhu, G.; Foletti, D.; Liu, X.; Ding, S.; Witt, J.M.; Hasa-Moreno, A.; Rickert, M.; Holz, C.; Aschenbrenner, L.; Yang, A.H.; et al. Targeting CLDN18.2 by CD3 Bispecific and ADC Modalities for the Treatments of Gastric and Pancreatic Cancer. Sci. Rep. 2019, 9, 8420.
- Yeung, Y.A.; Krishnamoorthy, V.; Dettling, D.; Sommer, C.; Poulsen, K.; Ni, I.; Pham, A.; Chen, W.; Liao-Chan, S.; Lindquist, K.; et al. An Optimized Full-Length FLT3/CD3 Bispecific Antibody Demonstrates Potent Anti-leukemia Activity and Reversible Hematological Toxicity. Mol. Ther. 2020, 28, 889–900.
- Wu, C.; Ying, H.; Grinnell, C.; Bryant, S.; Miller, R.; Clabbers, A.; Bose, S.; McCarthy, D.; Zhu, R.R.; Santora, L.; et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007, 25, 1290–1297.
- Bardwell, P.D.; Staron, M.M.; Liu, J.; Tao, Q.; Scesney, S.; Bukofzer, G.; Rodriguez, L.E.; Choi, C.H.; Wang, J.; Chang, Q.; et al. Potent and conditional redirected T cell killing of tumor cells using Half DVD-Ig. Protein Cell 2018, 9, 121–129.
- Correnti, C.E.; Laszlo, G.S.; de van der Schueren, W.J.; Godwin, C.D.; Bandaranayake, A.; Busch, M.A.; Gudgeon, C.J.; Bates, O.M.; Olson, J.M.; Mehlin, C.; et al. Simultaneous multiple interaction T-cell engaging (SMITE) bispecific antibodies overcome bispecific T-cell engager (BiTE) resistance via CD28 co-stimulation. Leukemia 2018, 32, 1239–1243.
- Steinmetz, A.; Vallée, F.; Beil, C.; Lange, C.; Baurin, N.; Beninga, J.; Capdevila, C.; Corvey, C.; Dupuy, A.; Ferrari, P.; et al. CODV-Ig, a universal bispecific tetravalent and multifunctional immunoglobulin format for medical applications. MAbs 2016, 8, 867–878.
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245.
- Lambert, J.M.; Berkenblit, A. Antibody-Drug Conjugates for Cancer Treatment. Annu. Rev. Med. 2018, 69, 191–207.
- Neri, D. Antibody-Cytokine Fusions: Versatile Products for the Modulation of Anticancer Immunity. Cancer Immunol. Res. 2019, 7, 348–354.
- Pardridge, W.M. Delivery of Biologics across the Blood-Brain Barrier with Molecular Trojan Horse Technology. BioDrugs 2017, 31, 503–519.
- Yu, Y.J.; Atwal, J.K.; Zhang, Y.; Tong, R.K.; Wildsmith, K.R.; Tan, C.; Bien-Ly, N.; Hersom, M.; Maloney, J.A.; Meilandt, W.J.; et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci. Transl. Med. 2014, 6, 261ra154.
- Yu, Y.J.; Zhang, Y.; Kenrick, M.; Hoyte, K.; Luk, W.; Lu, Y.; Atwal, J.; Elliott, J.M.; Prabhu, S.; Watts, R.J.; et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 2011, 3, 84ra44.
- Sampei, Z.; Igawa, T.; Soeda, T.; Okuyama-Nishida, Y.; Moriyama, C.; Wakabayashi, T.; Tanaka, E.; Muto, A.; Kojima, T.; Kitazawa, T.; et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS ONE 2013, 8, e57479.
- Kitazawa, T.; Shima, M. Emicizumab, a humanized bispecific antibody to coagulation factors IXa and X with a factor VIIIa-cofactor activity. Int. J. Hematol. 2020, 111, 20–30.
- Neiveyans, M.; Melhem, R.; Arnoult, C.; Bourquard, T.; Jarlier, M.; Busson, M.; Laroche, A.; Cerutti, M.; Pugnière, M.; Ternant, D.; et al. A recycling anti-transferrin receptor-1 monoclonal antibody as an efficient therapy for erythroleukemia through target up-regulation and antibody-dependent cytotoxic effector functions. MAbs 2019, 11, 593–605.
- Baruch, A.; Wong, C.; Chinn, L.W.; Vaze, A.; Sonoda, J.; Gelzleichter, T.; Chen, S.; Lewin-Koh, N.; Morrow, L.; Dheerendra, S.; et al. Antibody-mediated activation of the FGFR1/Klothobeta complex corrects metabolic dysfunction and alters food preference in obese humans. Proc. Natl. Acad. Sci. USA 2020, 117, 28992–29000.
- Shi, S.Y.; Lu, Y.W.; Liu, Z.; Stevens, J.; Murawsky, C.M.; Wilson, V.; Hu, Z.; Richards, W.G.; Michaels, M.L.; Zhang, J.; et al. A biparatopic agonistic antibody that mimics fibroblast growth factor 21 ligand activity. J. Biol. Chem. 2018, 293, 5909–5919.
- Gumlaw, N.; Sevigny, L.M.; Zhao, H.; Luo, Z.; Bangari, D.S.; Masterjohn, E.; Chen, Y.; McDonald, B.; Magnay, M.; Travaline, T.; et al. biAb Mediated Restoration of the Linkage between Dystroglycan and Laminin-211 as a Therapeutic Approach for alpha-Dystroglycanopathies. Mol. Ther. 2020, 28, 664–676.
- Fernandes, R.A.; Su, L.; Nishiga, Y.; Ren, J.; Bhuiyan, A.M.; Cheng, N.; Kuo, C.J.; Picton, L.K.; Ohtsuki, S.; Majzner, R.G.; et al. Immune receptor inhibition through enforced phosphatase recruitment. Nature 2020, 586, 779–784.
- Kontermann, R.E. Half-life extended biotherapeutics. Expert Opin. Biol. Ther. 2016, 16, 903–915.
- Schellenberger, V.; Wang, C.W.; Geething, N.C.; Spink, B.J.; Campbell, A.; To, W.; Scholle, M.D.; Yin, Y.; Yao, Y.; Bogin, O.; et al. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat. Biotechnol. 2009, 27, 1186–1190.
- Hutt, M.; Färber-Schwarz, A.; Unverdorben, F.; Richter, F.; Kontermann, R.E. Plasma half-life extension of small recombinant antibodies by fusion to immunoglobulin-binding domains. J. Biol. Chem. 2012, 287, 4462–4469.
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876.
- Fan, G.; Wang, Z.; Hao, M.; Li, J. Bispecific antibodies and their applications. J. Hematol. Oncol. 2015, 8, 130.
- Chari, R.V.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. Engl. 2014, 53, 3796–3827.
- Sellmann, C.; Doerner, A.; Knuehl, C.; Rasche, N.; Sood, V.; Krah, S.; Rhiel, L.; Messemer, A.; Wesolowski, J.; Schuette, M.; et al. Balancing Selectivity and Efficacy of Bispecific Epidermal Growth Factor Receptor (EGFR) x c-MET Antibodies and Antibody-Drug Conjugates. J. Biol. Chem. 2016, 291, 25106–25119.
- Wagner-Rousset, E.; Janin-Bussat, M.C.; Colas, O.; Excoffier, M.; Ayoub, D.; Haeuw, J.F.; Rilatt, I.; Perez, M.; Corvaïa, N.; Beck, A. Antibody-drug conjugate model fast characterization by LC-MS following IdeS proteolytic digestion. MAbs 2014, 6, 273–285.
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803.
- Levin, A.A. Treating Disease at the RNA Level with Oligonucleotides. N. Engl. J. Med. 2019, 380, 57–70.
- Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release 2016, 237, 1–13.
- Satake, N.; Duong, C.; Yoshida, S.; Oestergaard, M.; Chen, C.; Peralta, R.; Guo, S.; Seth, P.P.; Li, Y.; Beckett, L.; et al. Novel Targeted Therapy for Precursor B Cell Acute Lymphoblastic Leukemia: Anti-CD22 Antibody-MXD3 Antisense Oligonucleotide Conjugate. Mol. Med. 2016, 22, 632–642.
- Arnold, A.E.; Malek-Adamian, E.; Le, P.U.; Meng, A.; Martínez-Montero, S.; Petrecca, K.; Damha, M.J.; Shoichet, M.S. Antibody-Antisense Oligonucleotide Conjugate Downregulates a Key Gene in Glioblastoma Stem Cells. Mol. Ther. Nucleic Acids 2018, 11, 518–527.
- Erickson, H.K.; Park, P.U.; Widdison, W.C.; Kovtun, Y.V.; Garrett, L.M.; Hoffman, K.; Lutz, R.J.; Goldmacher, V.S.; Blättler, W.A. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006, 66, 4426–4433.
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374.
- Acchione, M.; Kwon, H.; Jochheim, C.M.; Atkins, W.M. Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody-drug conjugates. MAbs 2012, 4, 362–372.
- Sun, M.M.; Beam, K.S.; Cerveny, C.G.; Hamblett, K.J.; Blackmore, R.S.; Torgov, M.Y.; Handley, F.G.; Ihle, N.C.; Senter, P.D.; Alley, S.C. Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug. Chem. 2005, 16, 1282–1290.
- Junutula, J.R.; Gerber, H.P. Next-Generation Antibody-Drug Conjugates (ADCs) for Cancer Therapy. ACS Med. Chem. Lett. 2016, 7, 972–973.
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014, 6, 34–45.
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932.
- Shen, B.Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.F.; Mai, E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189.
- Kung Sutherland, M.S.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, I.; Ryan, M.C.; Sussman, D.; Lyon, R.P.; et al. SGN-CD33A: A novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 2013, 122, 1455–1463.
- Zhong, X.; He, T.; Prashad, A.S.; Wang, W.; Cohen, J.; Ferguson, D.; Tam, A.S.; Sousa, E.; Lin, L.; Tchistiakova, L.; et al. Mechanistic understanding of the cysteine capping modifications of antibodies enables selective chemical engineering in live mammalian cells. J. Biotechnol. 2017, 248, 48–58.
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106.
- Strop, P.; Liu, S.H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.T.; Ho, W.H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location matters: Site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 2013, 20, 161–167.
- Lhospice, F.; Brégeon, D.; Belmant, C.; Dennler, P.; Chiotellis, A.; Fischer, E.; Gauthier, L.; Boëdec, A.; Rispaud, H.; Savard-Chambard, S.; et al. Site-Specific Conjugation of Monomethyl Auristatin E to Anti-CD30 Antibodies Improves Their Pharmacokinetics and Therapeutic Index in Rodent Models. Mol. Pharm. 2015, 12, 1863–1871.
- Beerli, R.R.; Hell, T.; Merkel, A.S.; Grawunder, U. Sortase Enzyme-Mediated Generation of Site-Specifically Conjugated Antibody Drug Conjugates with High In Vitro and In Vivo Potency. PLoS ONE 2015, 10, e0131177.
- van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native mAbs Provides Homogeneous and Highly Efficacious Antibody-Drug Conjugates. Bioconjug. Chem. 2015, 26, 2233–2242.
- Rabuka, D.; Rush, J.S.; Gregory, W.D.; Wu, P.; Bertozzi, C.R. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc. 2012, 7, 1052–1067.
- Lin, S.; Yang, X.; Jia, S.; Weeks, A.M.; Hornsby, M.; Lee, P.S.; Nichiporuk, R.V.; Iavarone, A.T.; Wells, J.A.; Toste, F.D.; et al. Redox-based reagents for chemoselective methionine bioconjugation. Science 2017, 355, 597–602.
- Elledge, S.K.; Tran, H.L.; Christian, A.H.; Steri, V.; Hann, B.; Toste, F.D.; Chang, C.J.; Wells, J.A. Systematic identification of engineered methionines and oxaziridines for efficient, stable, and site-specific antibody bioconjugation. Proc. Natl. Acad. Sci. USA 2020, 117, 5733–5740.
- Mäger, I.; Meyer, A.H.; Li, J.; Lenter, M.; Hildebrandt, T.; Leparc, G.; Wood, M.J. Targeting blood-brain-barrier transcytosis—Perspectives for drug delivery. Neuropharmacology 2017, 120, 4–7.
- Niewoehner, J.; Bohrmann, B.; Collin, L.; Urich, E.; Sade, H.; Maier, P.; Rueger, P.; Stracke, J.O.; Lau, W.; Tissot, A.C.; et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 2014, 81, 49–60.
- Bezabeh, B.; Fleming, R.; Fazenbaker, C.; Zhong, H.; Coffman, K.; Yu, X.Q.; Leow, C.C.; Gibson, N.; Wilson, S.; Stover, C.K.; et al. Insertion of scFv into the hinge domain of full-length IgG1 monoclonal antibody results in tetravalent bispecific molecule with robust properties. MAbs 2017, 9, 240–256.
- Wec, A.Z.; Nyakatura, E.K.; Herbert, A.S.; Howell, K.A.; Holtsberg, F.W.; Bakken, R.R.; Mittler, E.; Christin, J.R.; Shulenin, S.; Jangra, R.K.; et al. A “Trojan horse” bispecific-antibody strategy for broad protection against ebolaviruses. Science 2016, 354, 350–354.
- Desnoyers, L.R.; Vasiljeva, O.; Richardson, J.H.; Yang, A.; Menendez, E.E.; Liang, T.W.; Wong, C.; Bessette, P.H.; Kamath, K.; Moore, S.J.; et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci. Transl. Med. 2013, 5, 207ra144.
- Mimoto, F.; Tatsumi, K.; Shimizu, S.; Kadono, S.; Haraya, K.; Nagayasu, M.; Suzuki, Y.; Fujii, E.; Kamimura, M.; Hayasaka, A.; et al. Exploitation of Elevated Extracellular ATP to Specifically Direct Antibody to Tumor Microenvironment. Cell Rep. 2020, 33, 108542.
- Kamata-Sakurai, M.; Narita, Y.; Hori, Y.; Nemoto, T.; Uchikawa, R.; Honda, M.; Hironiwa, N.; Taniguchi, K.; Shida-Kawazoe, M.; Metsugi, S.; et al. Antibody to CD137 Activated by Extracellular Adenosine Triphosphate Is Tumor Selective and Broadly Effective In Vivo without Systemic Immune Activation. Cancer Discov. 2021, 11, 158–175.
- Polu, K.R.; Lowman, H.B. Probody therapeutics for targeting antibodies to diseased tissue. Expert Opin. Biol. Ther. 2014, 14, 1049–1053.
- Maniaci, C.; Ciulli, A. Bifunctional chemical probes inducing protein-protein interactions. Curr. Opin. Chem. Biol. 2019, 52, 145–156.
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27.
- Verma, R.; Mohl, D.; Deshaies, R.J. Harnessing the Power of Proteolysis for Targeted Protein Inactivation. Mol. Cell. 2020, 77, 446–460.
- Kanner, S.A.; Morgenstern, T.; Colecraft, H.M. Sculpting ion channel functional expression with engineered ubiquitin ligases. Elife 2017, 6, e29744.
- Kanner, S.A.; Shuja, Z.; Choudhury, P.; Jain, A.; Colecraft, H.M. Targeted deubiquitination rescues distinct trafficking-deficient ion channelopathies. Nat. Methods 2020, 17, 1245–1253.
- Grabulovski, D.; Kaspar, M.; Neri, D. A novel, non-immunogenic Fyn SH3-derived binding protein with tumor vascular targeting properties. J. Biol. Chem. 2007, 282, 3196–3204.
- Silacci, M.; Lembke, W.; Woods, R.; Attinger-Toller, I.; Baenziger-Tobler, N.; Batey, S.; Santimaria, R.; von der Bey, U.; Koenig-Friedrich, S.; Zha, W.; et al. Discovery and characterization of COVA322, a clinical-stage bispecific TNF/IL-17A inhibitor for the treatment of inflammatory diseases. MAbs 2016, 8, 141–149.
- Bloom, L.; Calabro, V. FN3: A new protein scaffold reaches the clinic. Drug Discov. Today 2009, 14, 949–955.
- Koide, A.; Wojcik, J.; Gilbreth, R.N.; Hoey, R.J.; Koide, S. Teaching an old scaffold new tricks: Monobodies constructed using alternative surfaces of the FN3 scaffold. J. Mol. Biol. 2012, 415, 393–405.
- Koide, A.; Bailey, C.W.; Huang, X.; Koide, S. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 1998, 284, 1141–1151.
- Diem, M.D.; Hyun, L.; Yi, F.; Hippensteel, R.; Kuhar, E.; Lowenstein, C.; Swift, E.J.; O’Neil, K.T.; Jacobs, S.A. Selection of high-affinity Centyrin FN3 domains from a simple library diversified at a combination of strand and loop positions. Protein Eng. Des. Sel. 2014, 27, 419–429.
- Wojcik, J.; Hantschel, O.; Grebien, F.; Kaupe, I.; Bennett, K.L.; Barkinge, J.; Jones, R.B.; Koide, A.; Superti-Furga, G.; Koide, S. A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat. Struct. Mol. Biol. 2010, 17, 519–527.
- Ludwicki, M.B.; Li, J.; Stephens, E.A.; Roberts, R.W.; Koide, S.; Hammond, P.T.; DeLisa, M.P. Broad-Spectrum Proteome Editing with an Engineered Bacterial Ubiquitin Ligase Mimic. ACS Cent. Sci. 2019, 5, 852–866.
- Goldberg, S.D.; Cardoso, R.M.; Lin, T.; Spinka-Doms, T.; Klein, D.; Jacobs, S.A.; Dudkin, V.; Gilliland, G.; O’Neil, K.T. Engineering a targeted delivery platform using Centyrins. Protein Eng. Des. Sel. 2016, 29, 563–572.
- Feldwisch, J.; Tolmachev, V. Engineering of affibody molecules for therapy and diagnostics. Methods Mol. Biol. 2012, 899, 103–126.
- Löfblom, J.; Feldwisch, J.; Tolmachev, V.; Carlsson, J.; Ståhl, S.; Frejd, F.Y. Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010, 584, 2670–2680.
- Frejd, F.Y.; Kim, K.T. Affibody molecules as engineered protein drugs. Exp. Mol. Med. 2017, 49, e306.
- Akhtari, J.; Rezayat, S.M.; Teymouri, M.; Alavizadeh, S.H.; Gheybi, F.; Badiee, A.; Jaafari, M.R. Targeting, bio distributive and tumor growth inhibiting characterization of anti-HER2 affibody coupling to liposomal doxorubicin using BALB/c mice bearing TUBO tumors. Int. J. Pharm. 2016, 505, 89–95.
- Meister, S.W.; Hjelm, L.C.; Dannemeyer, M.; Tegel, H.; Lindberg, H.; Ståhl, S.; Löfblom, J. An Affibody Molecule Is Actively Transported into the Cerebrospinal Fluid via Binding to the Transferrin Receptor. Int. J. Mol. Sci. 2020, 21, 2999.
- Pluckthun, A. Designed ankyrin repeat proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511.
- Simon, M.; Stefan, N.; Borsig, L.; Plückthun, A.; Zangemeister-Wittke, U. Increasing the antitumor effect of an EpCAM-targeting fusion toxin by facile click PEGylation. Mol. Cancer Ther. 2014, 13, 375–385.
- Sokolova, E.; Proshkina, G.; Kutova, O.; Shilova, O.; Ryabova, A.; Schulga, A.; Stremovskiy, O.; Zdobnova, T.; Balalaeva, I.; Deyev, S. Recombinant targeted toxin based on HER2-specific DARPin possesses a strong selective cytotoxic effect in vitro and a potent antitumor activity in vivo. J. Control. Release 2016, 233, 48–56.
- Soto-Gamez, A.; Chen, D.; Nabuurs, A.G.; Quax, W.J.; Demaria, M.; Boersma, Y.L. A Bispecific Inhibitor of the EGFR/ADAM17 Axis Decreases Cell Proliferation and Migration of EGFR-Dependent Cancer Cells. Cancers 2020, 12, 411.
- Baird, R.; Linossi, C.; Middleton, M.; Lord, S.; Harris, A.; Rodón, J.; Zitt, C.; Fiedler, U.; Dawson, K.M.; Leupin, N.; et al. First-in-Human Phase I Study of MP0250, a First-in-Class DARPin Drug Candidate Targeting VEGF and HGF, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2020, JCO2000596.
- Babaee, N.; Garoosi, Y.T.; Karimipoor, M.; Davami, F.; Bayat, E.; Safarpour, H.; Mahboudi, F.; Barkhordari, F. DARPin Ec1-LMWP protein scaffold in targeted delivery of siRNA molecules through EpCAM cancer stem cell marker. Mol. Biol. Rep. 2020, 47, 7323–7331.
- Cox, N.; Kintzing, J.R.; Smith, M.; Grant, G.A.; Cochran, J.R. Integrin-Targeting Knottin Peptide-Drug Conjugates Are Potent Inhibitors of Tumor Cell Proliferation. Angew. Chem. Int. Ed. Engl. 2016, 55, 9894–9897.
- Lee, S.C.; Park, K.; Han, J.; Lee, J.J.; Kim, H.J.; Hong, S.; Heu, W.; Kim, Y.J.; Ha, J.S.; Lee, S.G.; et al. Design of a binding scaffold based on variable lymphocyte receptors of jawless vertebrates by module engineering. Proc. Natl. Acad. Sci. USA 2012, 109, 3299–3304.
- Richter, A.; Eggenstein, E.; Skerra, A. Anticalins: Exploiting a non-Ig scaffold with hypervariable loops for the engineering of binding proteins. FEBS Lett. 2014, 588, 213–218.
- Rothe, C.; Skerra, A. Anticalin((R)) Proteins as Therapeutic Agents in Human Diseases. BioDrugs 2018, 32, 233–243.
- Thakur, A.; Lum, L.G. Cancer therapy with bispecific antibodies: Clinical experience. Curr. Opin. Mol. Ther. 2010, 12, 340–349.
- Doppalapudi, V.R.; Huang, J.; Liu, D.; Jin, P.; Liu, B.; Li, L.; Desharnais, J.; Hagen, C.; Levin, N.J.; Shields, M.J.; et al. Chemical generation of bispecific antibodies. Proc. Natl. Acad. Sci. USA 2010, 107, 22611–22616.
- White, B.H.; Whalen, K.; Kriksciukaite, K.; Alargova, R.; Au Yeung, T.; Bazinet, P.; Brockman, A.; DuPont, M.; Oller, H.; Lemelin, C.A.; et al. Discovery of an SSTR2-Targeting Maytansinoid Conjugate (PEN-221) with Potent Activity in Vitro and in Vivo. J. Med. Chem. 2019, 62, 2708–2719.
- Wang, L.; Xu, J.; Kong, Y.; Liang, R.; Li, W.; Li, J.; Lu, J.; Dimitrov, D.S.; Yu, F.; Wu, Y.; et al. Engineering a Novel Antibody-Peptide Bispecific Fusion Protein against MERS-CoV. Antibodies 2019, 8, 53.

