 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Camilla Russo | + 2996 word(s) | 2996 | 2021-04-20 07:51:21 | | | |
| 2 | Dean Liu | + 2986 word(s) | 2986 | 2021-04-20 08:02:34 | | |
Video Upload Options
Neurofibromatosis type 1 (NF1), the most frequent phakomatosis and one of the most common inherited tumor predisposition syndromes, is characterized by several manifestations that pervasively involve central and peripheral nervous system structures.
1. Introduction
Neurofibromatosis type 1 (NF1), the most frequent phakomatosis and one of the most common inherited tumor predisposition syndromes, is a multi-organ autosomal dominant disease with an incidence ranging between 1:2000 and 1:3000 newborns and a prevalence of about 1:4500 [1]. Clinical phenotype encompasses a wide range of manifestations specially involving the skin, nervous system, skeleton and vessels. Very few genotype–phenotype correlations have been identified; for the remaining cases, a broad phonotypical variability is reported among patients and even within the same family [2]. This variability can be, at least in part, ascribed to the inheritance pattern of mutation in the single causative gene NF1 (ch.17q22.1) encoding for the ubiquitous tumor suppressor protein neurofibromin. However, the existence of additional modifier genes has been hypothesized [3][4]. Neurofibromin controls cell-division cycle and differentiation by inactivation of the proto-oncogene KRAS. KRAS is a GTPase responsible for MAPK pathway upregulation, thus promoting cell proliferation, differentiation and migration by modulating the MEK/ERK and phosphatidylinositol 3-kinase—mammalian target of rapamycin signaling pathways. In NF1 patients, the heterozygous pathogenic gene variant is present in every nucleated cell; in case of loss of heterozygosity or when the second wild-type allele is inactivated by a new intra-genic mutation (according to the Knudson two-hit model), neurofibromin becomes inactive or lacking within the cell with repercussion on the RAS-MAPK pathway. Second-hit mutations in the neurofibromin encoding gene have been frequently demonstrated both in the case of NF1-associated tumors and in non-neoplastic NF1-related manifestations [5]. At present, more than 3000 different pathogenic variants of NF1 gene have been described, mostly represented by loss-of-function mutations (ranging from nonsense to missense mutations, from deletions to insertions, from frame-shifts to translocations) [6]. Moreover, although ubiquitous, NF1 is highly expressed in neural crest (NC) derived tissues where it has a prominent regulatory activity on neural stem cell proliferation and precursor migration, with a specific effect depending on the target cell type, arising from each segment of the neural tube (cephalic, vagal, trunk or sacral) [7] (Table 1).
Table 1. Neural crest (NC) cell populations’ localization, and major derivates during embryologic development.
| Location | Neural Crest-Derived Cells |
|---|---|
| Cranial NC | Chondrocytes, Osteocytes, Odontoblasts (Cranio-facial skeleton) Cranial ganglia, Thyroid cells |
| Vagal NC | Smooth muscle cells, Cardiac septa, Pericytes (Cardiac development) Enteric ganglia |
| Trunk NC | Schwann cells Pigmented cells (Melanocytes) Dorsal root ganglia, Sympathetic ganglia Adrenal medulla |
| Sacral NC | Sympathetic ganglia, Enteric ganglia |
Therefore, it is easy to understand why neuroectoderm-derived lineage cells are the most involved cells in NF1, and why the main disease-related features affect the central nervous system (CNS), peripheral nervous system (PNS) and skin. In spite of a better comprehension of the impact of neurofibromin deregulation on the RAS-MAPK pathway in tumorigenesis (the major cause of reduced life quality and expectancy), the role of NF1 mutation in non-tumor manifestations and its impact on phenotype are less clear at present.
2. Brain
2.1. Focal Abnormal Signal Intensities: Neuroradiological Tips and Tricks
Focal abnormal signal intensities (FASIs), sometimes referred to as unidentified bright objects (UBOs), are focal or diffuse areas of increased T2w signal intensity within brain tissue on magnetic resonance imaging (MRI). Found in about 90% of lifelong NF1 patients, these alterations are most frequently detected in the cerebellum, brainstem and basal ganglia (Figure 1); however, hemispheric and hippocampal lesions may appear over time, suggesting a different pathogenic mechanism compared to other localizations [8][9][10].
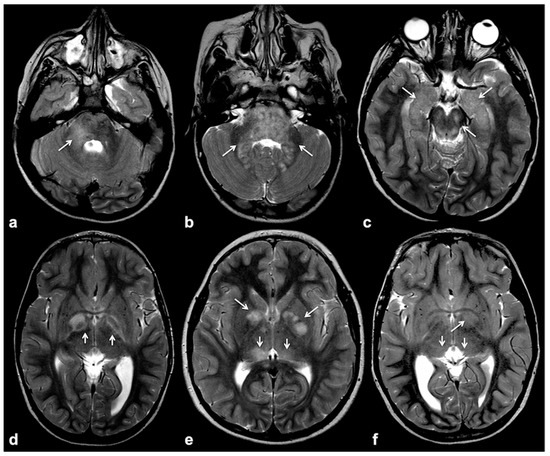
Figure 1. Typical focal abnormal signal intensity (FASI) localization to cerebellar pedicles and cerebellum (a,b), brainstem, cerebral peduncles and hippocampi (b,c), as well as basal ganglia (d–f) on axial TSE T2w MR images (white arrows).
FASIs vary in number and size over time, with the highest lesion burden detected in early childhood and the fastest decline observed in the thalami and cerebellum; hemispheric and deep FASIs tend to decrease in number with age, whereas diffuse lesions seem to be more stable over time [11][12][13][14]. Even if the sensitivity of FASI presence in NF1 is high, today they have just been proposed (and not yet accepted) as additional diagnostic criteria due to their relatively low specificity and variable localization; indeed, sensitivity and specificity of FASIs for NF1 averaged 97% and 79%, respectively [15]. In the only literature report of histologic examination, FASIs corresponded to areas of abnormally increased white matter volume corresponding to spongiform myelopathy with myelin vacuolization, but neither marked demyelination nor an inflammatory response [16][17]. These vacuolar changes due to intra-myelinic edema represent the major pathologic finding and could, at least in part, explain their MRI signal. At the MRI examination, FASIs are hyperintense on T2w images and iso-hyperintense on T1w, with no mass effect or post-contrast enhancement. It should be noticed that hyperintensity on T1w is generally limited to FASIs within the basal ganglia, and far less common in the posterior fossa and cerebral white matter, thus suggesting a difference in these localizations [18]; among possible causes of T1-shortening in basal ganglia foci, the most accredited theory is the presence of subtle microcalcification, as documented in previous reports. A possible explanation to this evolution in T1w signal is that calcifications are a late phenomenon due to reparative mechanisms to intra-myelinic edema [16][18].
If, in children, FASIs can be easily detected on turbo spin echo (TSE) T2w, in adult patients, fluid attenuation inversion recovery (FLAIR) and proton density (PD) sequences show higher sensitivity; these lesions are generally isointense on T1w and conventional diffusion weighted imaging (DWI), with a sometimes slight elevation in apparent diffusion coefficient values [19]. In rare cases, FASIs can also show atypical MRI features, such as a mild mass effect and focal contrast enhancement (generally transient and regressing over time) (Figure 2); being that these latter features are more typical of brain gliomas, a differential diagnosis with glial tumors can be very challenging.
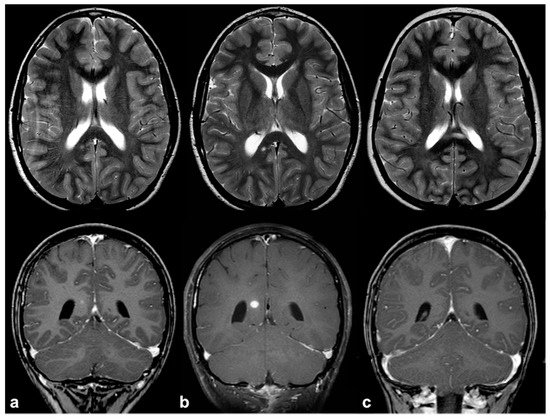
Figure 2. Longitudinal follow-up of atypical brain FASI in a 5 year-old NF1 male patient: axial TSE T2w (first row) and coronal post-contrast T1w (second row) MRI showing a oval-shaped lesion into the right occipital forceps of the splenium corpus callosum at three different time points (a = 5, b = 5.5, and c = 6 years old). The lesion was T2-hyperintense with punctate enhancement at diagnosis, showed a transient volume increase with focal contrast enhancement, and then completely regressed at one-year longitudinal MRI follow-up.
In this light, advanced techniques can help in the differential diagnosis from glial tumors [9][13][20]. Perfusion-weighted MRI may allow for the detection of tumor-related angiogenesis and increased vascular permeability, features that are usually absent in the case of FASIs and not always present in the case of low-grade glioma. Conversely, spectroscopy has the potential for distinguishing glial tumors (increased choline peak, decreased creatine peak, and absent N-acetylaspartate) from FASIs (where N-acetylaspartate levels are preserved). These spectral abnormalities are also visible in other regions involved in myelin vacuolization, even in absence of areas of patent hyperintensity [21][22]. These findings are further corroborated by microstructural studies, which (in spite of generally silent conventional DWI findings) highlight subtle alterations in fractional anisotropy and radial diffusivity consistent with intramyelin edema; these alterations can persist even after FASI regression [10][12][23][24]. From a prognostic viewpoint, FASIs are benign lesions; in spite of previous evidences suggesting a possible correlation between FASIs’ burden and cognitive impairment, recent studies concluded that they do not represent a strong indicator of executive dysfunction in children with NF1 [14][25]. Indeed, despite conflicting results from different studies [26], to date, FASIs are thought to play a marginal role in microstructural disruption and cognition; conversely, they should rather be considered as a late epiphenomenon of an underlying structural neurodevelopmental disorder [12][23][27]. This consideration may also explain why children with a higher NF1-associated lesion burden seem to have a worse clinical outcome compared to those with a lower lesion burden [14][25][28][29]. Finally, it should be noticed that, despite the fact that clinical management should not be affected by FASIs or optic nerve thickening in asymptomatic patients, their presence frequently results in closer clinical and MRI monitoring (with subsequent unbeneficial effects in terms of cost-effectiveness, as well as emotional burden for families).
2.2. Volume, Structural and Functional Connectivity Changes: More Than Meet the Eye
NF1 is characterized by a global increase in brain volume, sometimes associated with macrocephaly, more evident in white rather than in grey matter; these changes seem to be age-dependent and more pronounced in younger patients [17][30][31][32][33]. A significant volume increase is observed in midline structures, with particular reference to the corpus callosum which looks altered both in terms of micro- and macro-structural measurements [31][34]. Subcortical structures, such as the hippocampus, amygdala and basal ganglia (i.e., thalamus and striatum), can also present larger volumes than normal [17][35][36][37][38]. Conversely, grey matter density is lower in the frontal parietal and temporal lobes (and, to a lesser extent, in cingulate and insular regions) with simplified cortical gyration and abnormal cortical thickness that decreases with age [12][17][39][40]. From a micro-structural point of view, these volumetric changes correspond to an extensive global and local white matter disruption, as documented with diffusion tensor imaging (DTI) and a diffusion parameters analysis; indeed, several studies have documented an increased radial diffusivity with reduced fractional anisotropy in lobar white matter, suggesting a link between an impaired microstructure and abnormal fluid accumulation, potentially due to subtle myelin vacuolization [41][42][43]. Focal alterations are more evident in the frontal lobe white matter, [42] and in the anterior thalamic radiation, a white matter bundle connecting the thalamus with frontal lobes that are associated with higher executive functioning [27]. It should be noted that white matter disruption is observed independently from FASIs and other macroscopic NF1-associated lesions, supporting the thesis that axonal degeneration might occur even before, or in the absence of, primary myelination changes [43].
Several functional MRI (fMRI) studies have explored the functional equivalent of the above-described white matter disruption, with particular regard to the evaluation of the more compromised functions in NF1 (namely, executive and visuo-spatial). Both static functional connectivity and resting-state fMRI dynamic properties seemed to be affected in NF1 patients. Alterations in executive functioning correspond to a dysfunction in connectivity through the frontal, superior temporal, parietal and anterior cingulate cortex, with an extensive involvement of the motor, pre-motor and supplementary motor cortex [40][44][45][46]; similarly, for visuo-spatial functioning, a deficient activation of the low-level visual cortex due to anomalous and persistent activation of the default mode network during visual stimulation was observed [47]. Along with these static alterations, the dynamic properties of whole-brain connectivity at the resting-state fMRI were also reduced in NF1 patients compared to healthy controls. These findings are consistent with an overall reduction in the inhibitor neurotransmitter gamma-aminobutyric acid (GABA) levels, both in the occipital and frontal lobes, as documented by few MRI spectroscopy studies [48][49].
The clinical counterparty of the described widespread alterations is represented by the cognitive and behavioral deficits, whose severity may be poorly predicted by resorting to neuroimaging, despite the always more remarkable contribution of advanced MRI techniques. Coordination disorders with reduced visuo-spatial/motor abilities, comprehension deficit, linguistic impairment, autistic mannerisms and attention deficit/hyperactivity disorder represent the most common cognitive manifestations of NF1 [34][35][36][50]. All these findings taken together could probably reflect a delayed or aberrant dendritic pruning depending, at least in part, on NF1 gene mutations; at present, the correlation between impaired function and the involved brain regions is still poorly understood, although these preliminary findings suggest a possible role of these in vivo biomarkers for future disease monitoring and treatment response assessment [12]. However, the variable prevalence and penetrance of these cognitive dysfunctions in NF1 patients plead in favor of the existence of multiple physiopathological mechanisms, which cannot be fully elucidated by morphological and structural neuroimaging approaches alone [38].
2.3. Epileptogenic Lesions in NF1
NF1 is associated with higher seizure frequency compared to the general population, with a prevalence of about 5% (lifelong) and increasing incidence with age; clinical manifestations are usually represented by focal seizures onset with secondary generalization [51]. The majority of NF1 patients with seizures presented with focal epileptogenic lesions, and frequently with NF1-related tumors (about 60–65% of cases) [52][53]; however, mesial temporal sclerosis, malformations of cortical development or cerebrovascular lesions have also been sporadically reported as epileptogenic triggers accounting for about 20–25% of all cases [52][53][54], while no clear association is reported with FASI [52][54][55]. In the remaining cases, no focal brain lesion have been documented, and the exact cause of seizure onset in this NF1 patients subgroup is not yet clear [53]. The most reliable hypotheses suggest, as possible mechanisms, an excitation/inhibition imbalance in GABAergic signaling or an abnormal sensitization of ion channels mediated by the RAS-MAPK pathway [56][57][58]. In NF1, epilepsy is generally responsive to medical treatment, whereas in selected cases of medication-refractory, surgery may be envisaged to limit seizure recurrence; in these patients, pre-operative multimodal evaluation (combining electroencephalography, advanced/conventional MRI techniques, and nuclear medicine imaging) can be necessary to distinguish real epileptic trigger from non-epileptogenic lesions [54][59].
2.4. Altered Cerebrospinal Fluid Dynamics: Obstructive Hydrocephalus Other Than Tumor-Related
Hydrocephalus prevalence in NF1 ranges between 1% in adults and 13% in children, and it is almost invariably represented by non-communicating forms due to the impaired flow of CSF into the ventricular system [60]. Although frequently related to midbrain, diencephalon or basal ganglia masses, non-communicating hydrocephalus can be also due to non-neoplastic lesions. Among these causes of CSF flow alteration, aqueductal stenosis, aqueductal web and superior medullary velum synechiae, whose etiology still remains uncertain, have an increased incidence rate in patients with NF1 compared to the general population [61][62][63]. Symptoms vary depending on pathogenic cause and hydrocephalus entity, despite the fact that asymptomatic incidental dilatation of the ventricles can also be observed [60]. Once ruled out at the imaging of the presence of underlying neoplastic lesions, non-communicating hydrocephalus is generally treated by external shunting or endoscopic third-ventriculostomy (ETV). In the case of triventricular hydrocephalus, ETV represents the golden standard procedure with a high success rate and low risk of post-surgical complications; in some cases, the use of a trans-stoma stent may be required to ensure long-term stoma patency [60][64]. In this setting MRI plays a crucial role both at diagnosis and during the follow-up; besides conventional MRI, the use of 3D heavily T2-weighted sequences provides specific morphologic data regarding CSF pathways, thanks to high spatial resolution (Figure 3), whereas the application of phase-contrast techniques ensures the visualization of CSF dynamics through different compartments (also documenting the post-surgical patency of stoma/stent over time).
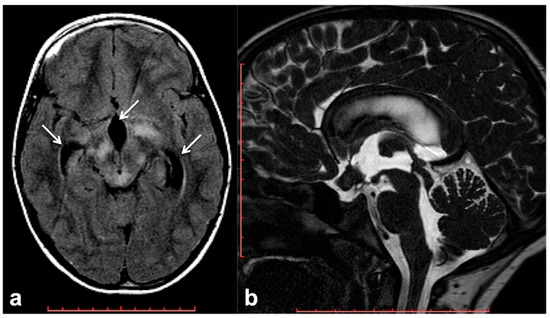
Figure 3. Brain MRI of a 6 year-old boy with NF1 and aqueductal stenosis: axial fluid attenuation inversion recovery (FLAIR) (a) showing bilateral basal ganglia and brainstem hyperintensities consistent with FASI. Supratentorial hydrocephalus with enlarged third ventricle and temporal horns of the lateral ventricles was also seen (white arrows); no transependymal CSF flow in periventricular areas was visible, and the fourth ventricle was normal. Sagittal 3D heavily T2-weighted sequence (b) revealed the presence of a thickened web in the cerebral aqueduct causing stenosis and hydrocephalus.
2.5. CNS Vascular Manifestations of NF1
CNS vascular abnormalities are an occasional finding in NF1 patients, with higher prevalence compared to the general population, ranging between 3% and 7% [65]. Cerebral arteriopathy mainly affects the arterial brain supply and, more specifically, the anterior circulation system. This is due to the embryologic origin of the internal carotid artery and its intracranial branches from the NC, whereas the posterior circulation ontogenetically arises from the paraxial mesoderm. Among possible cerebral vasculopathies, moyamoya syndrome (MMS) represents the most common finding, followed by cerebral vascular malformations and aneurysms [66][67][68][69] (Figure 4); all the vascular alterations are not strictly associated with other features of NF1 and do not correlate with disease severity [65].
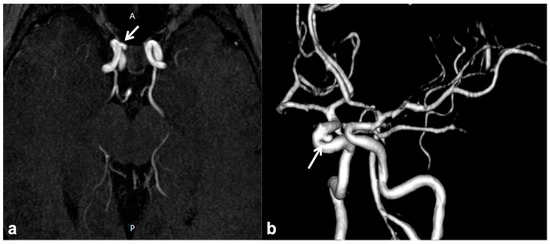
Figure 4. Non-contrast time-of-flight 3D MR angiography maximum intensity projection on the axial plane (a) and its volume rendering reconstruction (b) in a 10 year-old boy with NF1, showing the presence of a small saccular aneurysm of the right cavernous internal carotid artery (white arrows).
MMS is defined as a progressive cerebrovascular angiopathy in the setting of a predisposing condition, such as phakomatoses, caused by narrowing or stenosis in Willis’ polygon arteries, and manifesting as recurrent minor or major strokes. Symptoms are usually represented by acute-onset focal neurological deficits with sudden weakness or numbness in the face, arm or leg on one side; other possible manifestations also include headaches, visual disturbances, developmental delay and seizures [70]. Among phakomatoses, NF1 has been suggested as a possible MMS predisposing disorder, and few susceptibility genetic loci have been recently identified [37][71]. This is why, despite the fact that MMS was usually described as a consequence of cranial radiation therapy for NF1-related optic pathway glioma, the majority of cases have been reported as a primary manifestation of NF1 even in the absence of previous radiotherapy [65][72]. Despite their relatively high prevalence, these manifestations are frequently recognized as poorly symptomatic although potentially fatal; therefore, it may be worth using transcranial Doppler as a screening method for identifying cerebral vasculopathy in children with NF1 and always including unenhanced MR angiography in routine examination protocols of these patients (Figure 5) [73][74].
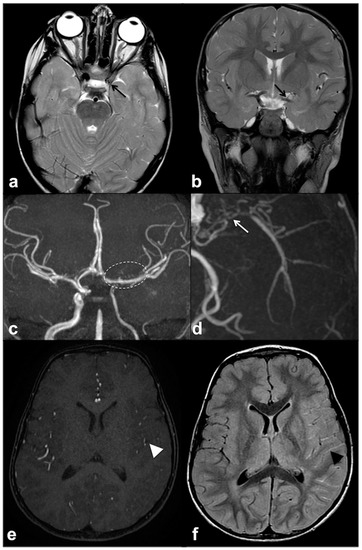
Figure 5. Axial (a) and coronal (b) TSE T2w MRI of an 8 year-old girl with NF1, showing reduced visualization of left cavernous-supraclinoid internal carotid and proximal left middle cerebral arteries’ flow void (black arrows). Time-of-flight 3D MR angiography maximum intensity projection (c) confirmed the severe stenosis of left internal carotid artery and proximal segment of left middle cerebral artery, whereas distal segments are partly supplied by collaterals; dotted line region magnification (d) showed multiple small collateral vessels in proximal left middle cerebral artery region (white arrow). Axial time-of-flight 3D MR angiography (e) confirmed a reduction in distal middle cerebral artery branches flow (white arrowhead), with a diffuse net of prominent leptomeningeal collaterals with high signal on FLAIR (f) due to slow flow (black arrowhead).
When clinical and radiological signs of MMS are detected, contrast-enhanced MRI perfusion techniques may represent a useful tool for grading hemodynamic insufficiency and determining the need for surgical revascularization [75]. Investigating vascular abnormalities by the above-mentioned non-invasive methods can help in preventing cerebrovascular complications and selecting patients for subsequent confirmation by more invasive techniques, such as digital subtraction angiography.
References
- Hirbe, A.C.; Gutmann, D.H. Neurofi bromatosis type 1: A multidisciplinary approach to care. Lancet Neurol. 2014, 13, 834–843.
- Jett, K.; Friedman, J.M. Clinical and genetic aspects of neurofibromatosis 1. Genet. Med. 2009, 12, 1–11.
- Cichowski, K.; Jacks, T. NF1 Tumor Suppressor Gene Function: Narrowing the GAP. Cell 2001, 104, 593–604.
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157.
- Legius, E.; Brems, H. Genetic basis of neurofibromatosis type 1 and related conditions, including mosaicism. Child’s Nerv. Syst. 2020, 36, 2285–2295.
- Koczkowska, M.; Callens, T.; Chen, Y.; Gomes, A.; Hicks, A.D.; Sharp, A.; Johns, E.; Uhas, K.A.; Armstrong, L.; Bosanko, K.A.; et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: Genotype–phenotype study in neurofibromatosis type 1. Hum. Mutat. 2019, 41, 299–315.
- Chen, Y.-H.; Gianino, S.M.; Gutmann, D.H. Neurofibromatosis-1 regulation of neural stem cell proliferation and multilineage differentiation operates through distinct RAS effector pathways. Genes Dev. 2015, 29, 1677–1682.
- Gill, D.S.; Hyman, S.L.; Steinberg, A.; North, K.N. Age-related findings on MRI in neurofibromatosis type 1. Pediatr. Radiol. 2006, 36, 1048–1056.
- Lopes Ferraz-Filho, J.R.; Munis, M.P.; Souza, A.S.; Sanches, R.A.; Goloni-Bertollo, E.M.; Pavarino-Bertelli, E.C. Unidentified bright objects on brain MRI in children as a diagnostic criterion for neurofibromatosis type 1. Pediatr. Radiol. 2008, 38, 305–310.
- Lopes Ferraz-Filho, R.J.; da Rocha, A.J.; Pontes Muniz, M.; Soares Souza, A.; Goloni-Bertollo, E.M.; Pavarino-Bertelli, E.C. Unidentified bright objects in neurofibromatosis type 1: Conventional MRI in the follow-up and correlation of microstructural lesions on diffusion tensor images. Eur. J. Paediatr. Neurol. 2012, 16, 42–47.
- Calvez, S.; Levy, R.; Calvez, R.; Roux, C.-J.; Grévent, D.; Purcell, Y.; Beccaria, K.; Blauwblomme, T.; Grill, J.; Dufour, C.; et al. Focal Areas of High Signal Intensity in Children with Neurofibromatosis Type 1: Expected Evolution on MRI. Am. J. Neuroradiol. 2020, 41, 1733–1739.
- Baudou, E.; Nemmi, F.; Biotteau, M.; Maziero, S.; Peran, P.; Chaix, Y. Can the Cognitive Phenotype in Neurofibromatosis Type 1 (NF1) Be Explained by Neuroimaging? A Review. Front. Neurol. 2020, 10, 1373.
- Salman, M.S.; Hossain, S.; Alqublan, L.; Bunge, M.; Rozovsky, K. Cerebellar radiological abnormalities in children with neurofibromatosis type 1: Part 1—Clinical and neuroimaging findings. Cerebellum Ataxias 2018, 5, 14.
- Salman, M.S.; Hossain, S.; Gorun, S.; Alqublan, L.; Bunge, M.; Rozovsky, K. Cerebellar radiological abnormalities in children with neurofibromatosis type 1: Part 2—A neuroimaging natural history study with clinical correlations. Cerebellum Ataxias 2018, 5, 13.
- DeBella, K.; Poskitt, K.; Szudek, J.; Friedman, J.M. Use of “unidentified bright objects” on MRI for diagnosis of neurofibromatosis 1 in children. Neurology 2000, 54, 1646–1651.
- Di Paolo, D.P.; Zimmerman, R.A.; Rorke, L.B.; Zackai, E.H.; Bilaniuk, L.T.; Yachnis, A.T. Neurofibromatosis type 1: Pathologic substrate of high-signal-intensity foci in the brain. Radiology 1995, 195, 721–724.
- Barkovich, M.J.; Tan, C.H.; Kline, C.; Dale, A.M.; Jernigan, T.L.; Sugrue, L.P.; Barkovich, A.J.; Desikan, R.S.; Nillo, R.M.; Li, Y.; et al. Abnormal Morphology of Select Cortical and Subcortical Regions in Neurofibromatosis Type 1. Radiology 2018, 289, 499–508.
- Terada, H.; Barkovich, A.J.; Edwards, M.S.; Ciricillo, S.M. Evolution of high-intensity basal ganglia lesions on T1-weighted MR in neurofibromatosis type 1. Am. J. Neuroradiol. 1996, 17, 755–760.
- Alkan, A.; Sigirci, A.; Kutlu, R.; Ozcan, H.; Erdem, G.; Aslan, M.; Ates, O.; Yakinci, C.; Egri, M. Neurofibromatosis type 1: Diffusion weighted imaging findings of brain. Eur. J. Radiol. 2005, 56, 229–234.
- Tritt, S.; Hillenbrand, N.; Liesirova, K.; Moein, G.; Kieslich, M.; Porto, L. Comparison of the detectability of UBOs in Neurofibromatosis Type I patients with proton density-weighted and FLAIR sequences in 3T MRI. Eur. J. Paediatr. Neurol. 2018, 22, 615–619.
- Wang, P.Y.; Kaufmann, W.E.; Koth, C.W.; Denckla, M.B.; Barker, P.B. Thalamic involvement in neurofibromatosis type 1: Evaluation with proton magnetic resonance spectroscopic imaging. Ann. Neurol. 2000, 47, 477–484.
- Barbier, C.; Chabernaud, C.; Barantin, L.; Bertrand, P.; Sembely, C.; Sirinelli, D.; Castelnau, P.; Cottier, J.-P. Proton MR spectroscopic imaging of basal ganglia and thalamus in neurofibromatosis type 1: Correlation with T2 hyperintensities. Neuroradiology 2010, 53, 141–148.
- Billiet, T.; Mädler, B.; D’Arco, F.; Peeters, R.; Deprez, S.; Plasschaert, E.; Leemans, A.; Zhang, H.; Bergh, B.V.D.; Vandenbulcke, M.; et al. Characterizing the microstructural basis of “unidentified bright objects” in neurofibromatosis type 1: A combined in vivo multicomponent T2 relaxation and multi-shell diffusion MRI analysis. NeuroImage Clin. 2014, 4, 649–658.
- Rosenbaum, T.; Engelbrecht, V.; Krölls, W.; Van Dorsten, F.A.; Hoehn-Berlage, M.; Lenard, H.-G. MRI abnormalities in neurofibromatosis type 1 (NF1): A study of men and mice. Brain Dev. 1999, 21, 268–273.
- Eby, N.S.; Griffith, J.L.; Gutmann, D.H.; Morris, S.M. Adaptive functioning in children with neurofibromatosis type 1: Relationship to cognition, behavior, and magnetic resonance imaging. Dev. Med. Child Neurol. 2019, 61, 972–978.
- Roy, A.; Barbarot, S.; Charbonnier, V.; Gayet-Delacroix, M.; Stalder, J.-F.; Roulin, J.-L.; Le Gall, D. Examining the frontal subcortical brain vulnerability hypothesis in children with neurofibromatosis type 1: Are T2-weighted hyperintensities related to executive dysfunction? Neuropsychology 2015, 29, 473–484.
- Koini, M.; Rombouts, S.A.R.B.; Veer, I.M.; Van Buchem, M.A.; Huijbregts, S.C.J. White matter microstructure of patients with neurofibromatosis type 1 and its relation to inhibitory control. Brain Imaging Behav. 2017, 11, 1731–1740.
- Goh, W.H.S.; Khong, P.-L.; Leung, C.S.Y.; Wong, V.C.N. T 2-Weighted Hyperintensities (Unidentified Bright Objects) in Children With Neurofibromatosis 1: Their Impact on Cognitive Function. J. Child Neurol. 2004, 19, 853–858.
- Chabernaud, C.; Sirinelli, D.; Barbier, C.; Cottier, J.-P.; Sembely, C.; Giraudeau, B.; Deseille-Turlotte, G.; Lorette, G.; Barthez, M.-A.; Castelnau, P. Thalamo-Striatal T2-Weighted Hyperintensities (Unidentified Bright Objects) Correlate With Cognitive Impairments in Neurofibromatosis Type 1 During Childhood. Dev. Neuropsychol. 2009, 34, 736–748.
- DiMario, F.J. Brain Abnormalities in Tuberous Sclerosis Complex. J. Child Neurol. 2004, 19, 650–657.
- Moore, B.D., III; Slopis, J.M.; Jackson, E.F.; De Winter, A.E.; Leeds, N.E. Brain volume in children with neurofibromatosis type 1. Relation to neuropsychological status. Neurology 2000, 54, 914–920.
- Giedd, J.N.; Castellanos, F.; Casey, B.J.; Kozuch, P.; King, A.C.; Hamburger, S.D.; Rapoport, J.L. Quantitative morphology of the corpus callosum in attention deficit hyperactivity disorder. Am. J. Psychiatry 1994, 151, 665–669.
- Cutting, L.; Cooper, K.; Koth, C.; Mostofsky, S.; Kates, W.; Denckla, M.; Kaufmann, W. Megalencephaly in NF1: Predominantly white matter contribution and mitigation by ADHD. Neurology 2002, 59, 1388–1394.
- Aydin, S.; Kurtcan, S.; Alkan, A.; Guler, S.; Filiz, M.; Yilmaz, T.F.; Sahin, T.U.; Aralasmak, A. Relationship between the corpus callosum and neurocognitive disabilities in children with NF-1: Diffusion tensor imaging features. Clin. Imaging 2016, 40, 1092–1095.
- Violante, I.R.; Ribeiro, M.J.; Silva, E.D.; Castelo-Branco, M. Gyrification, cortical and subcortical morphometry in neurofibromatosis type 1: An uneven profile of developmental abnormalities. J. Neurodev. Disord. 2013, 5, 3.
- Huijbregts, S.C.; Loitfelder, M.; Rombouts, S.A.; Swaab, H.; Verbist, B.M.; Arkink, E.B.; Van Buchem, M.A.; Veer, I.M. Cerebral volumetric abnormalities in Neurofibromatosis type 1: Associations with parent ratings of social and attention problems, executive dysfunction, and autistic mannerisms. J. Neurodev. Disord. 2015, 7, 1–9.
- Santoro, C.; Giugliano, T.; Bernardo, P.; Palladino, F.; Torella, A.; Blanco, F.D.V.; Onore, M.E.; Carotenuto, M.; Nigro, V.; Piluso, G. A novel RAB39B mutation and concurrent de novo NF1 mutation in a boy with neurofibromatosis type 1, intellectual disability, and autism: A case report. BMC Neurol. 2020, 20, 327.
- Baudou, E.; Nemmi, F.; Biotteau, M.; Maziero, S.; Assaiante, C.; Cignetti, F.; Vaugoyeau, M.; Audic, F.; Peran, P.; Chaix, Y. Are morphological and structural MRI characteristics related to specific cognitive impairments in neurofibromatosis type 1 (NF1) children? Eur. J. Paediatr. Neurol. 2020, 28, 89–100.
- Billingsley, R.L.; Schrimsher, G.W.; Jackson, E.F.; Slopis, J.M.; Moore, B.D., III. Significance of Planum Temporale and Planum Parietale Morphologic Features in Neurofibromatosis Type 1. Arch. Neurol. 2002, 59, 616–622.
- Billingsley, R.L.; Jackson, E.F.; Slopis, J.M.; Swank, P.R.; Mahankali, S.; Moore, B.D., III. Functional Magnetic Resonance Imaging of Phonologic Processing in Neurofibromatosis 1. J. Child. Neurol. 2003, 18, 731–740.
- Van Engelen, S.; Krab, L.C.; Moll, H.A.; de Goede-Bolder, A.; Pluijm, S.; Catsman-Berrevoets, C.; Elgersma, Y.; Lequin, M.H. Quantitative Differentiation Between Healthy and Disordered Brain Matter in Patients with Neurofibromatosis Type I Using Diffusion Tensor Imaging. Am. J. Neuroradiol. 2008, 29, 816–822.
- Nemmi, F.; Cignetti, F.; Assaiante, C.; Maziero, S.; Audic, F.; Péran, P.; Chaix, Y. Discriminating between neurofibromatosis-1 and typically developing children by means of multimodal MRI and multivariate analyses. Hum. Brain Mapp. 2019, 40, 3508–3521.
- Karlsgodt, K.H.; Rosser, T.; Lutkenhoff, E.S.; Cannon, T.D.; Silva, A.; Bearden, C.E. Alterations in White Matter Microstructure in Neurofibromatosis-1. PLoS ONE 2012, 7, e47854.
- Ibrahim, A.F.; Montojo, C.A.; Haut, K.M.; Karlsgodt, K.H.; Hansen, L.; Congdon, E.; Rosser, T.; Bilder, R.M.; Silva, A.J.; Bearden, C.E. Spatial working memory in neurofibromatosis 1: Altered neural activity and functional connectivity. NeuroImage Clin. 2017, 15, 801–811.
- Loitfelder, M.; Huijbregts, S.C.; Veer, I.M.; Swaab, H.S.; Van Buchem, M.A.; Schmidt, R.; Rombouts, S.A. Functional Connectivity Changes and Executive and Social Problems in Neurofibromatosis Type I. Brain Connect. 2015, 5, 312–320.
- Tomson, S.N.; Schreiner, M.J.; Narayan, M.; Rosser, T.; Enrique, N.; Silva, A.J.; Allen, G.I.; Bookheimer, S.Y.; Bearden, C.E. Resting state functional MRI reveals abnormal network connectivity in neurofibromatosis 1. Hum. Brain Mapp. 2015, 36, 4566–4581.
- Violante, I.R.; Ribeiro, M.J.; Cunha, G.; Bernardino, I.; Duarte, J.V.; Ramos, F.; Saraiva, J.; Silva, E.; Castelo-Branco, M. Abnormal Brain Activation in Neurofibromatosis Type 1: A Link between Visual Processing and the Default Mode Network. PLoS ONE 2012, 7, e38785.
- Ribeiro, M.; Violante, R.; Bernardino, I.; Edden, R.; Castelo-Branco, M. ScienceDirect Abnormal relationship between GABA, neurophysiology and impulsive behavior in neurofibromatosis type 1. Cortex 2014, 4, 194–208.
- Violante, I.R.; Ribeiro, M.J.; Edden, R.A.E.; Guimarães, P.; Bernardino, I.; Rebola, J.; Cunha, G.; Silva, E.; Castelo-Branco, M. GABA deficit in the visual cortex of patients with neurofibromatosis type 1: Genotype–phenotype correlations and functional impact. Brain 2013, 136, 918–925.
- Hachon, C.; Iannuzzi, S.; Chaix, Y. Behavioural and cognitive phenotypes in children with neurofibromatosis type 1 (NF1): The link with the neurobiological level. Brain Dev. 2011, 33, 52–61.
- Bernardo, P.; Santoro, C.; Rubino, A.; Mirone, G.; Cinalli, G. Epilepsy surgery in neurofibromatosis type 1: An overlooked therapeutic approach. Child’s Nerv. Syst. 2020, 36, 2909–2910.
- Pecoraro, A.; Arehart, E.; Gallentine, W.; Radtke, R.; Smith, E.; Pizoli, C.; Kansagra, S.; Abdelnour, E.; McLendon, R.; Mikati, M.A. Epilepsy & Behavior Epilepsy in neurofibromatosis type 1. Epilepsy Behav. 2017, 73, 137–141.
- Ostendorf, A.P.; Gutmann, D.H.; Weisenberg, J.L.Z. Epilepsy in individuals with neurofibromatosis type 1. Epilepsia 2013, 54, 1810–1814.
- Bernardo, P.; Cinalli, G.; Santoro, C. Epilepsy in NF1: A systematic review of the literature. Child’s Nerv. Syst. 2020, 36, 2333–2350.
- Hsieh, H.-Y.; Fung, H.-C.; Wang, C.-J.; Chin, S.-C.; Wu, T. Epileptic seizures in neurofibromatosis type 1 are related to intracranial tumors but not to neurofibromatosis bright objects. Seizure 2011, 20, 606–611.
- Moutal, A.; Dustrude, E.T.; Khanna, R. Sensitization of Ion Channels Contributes to Central and Peripheral Dysfunction in Neurofibromatosis Type 1. Mol. Neurobiol. 2017, 54, 3342–3349.
- Stafstrom, C.E.; Staedtke, V.; Comi, A.M. Epilepsy Mechanisms in Neurocutaneous Disorders: Tuberous Sclerosis Complex, Neurofibromatosis Type 1, and Sturge–Weber Syndrome. Front. Neurol. 2017, 8, 87.
- Serdaroglu, E.; Konuskan, B.; Oguz, K.K.; Gurler, G.; Yalnizoglu, D.; Anlar, B. Epilepsy in neurofibromatosis type 1: Diffuse cerebral dysfunction? Epilepsy Behav. 2019, 98, 6–9.
- Barba, C.; Jacques, T.; Kahane, P.; Polster, T.; Isnard, J.; Leijten, F.S.; Özkara, Ç.; Tassi, L.; Giordano, F.; Castagna, M.; et al. Epilepsy surgery in Neurofibromatosis Type 1. Epilepsy Res. 2013, 105, 384–395.
- Roth, J.; Constantini, S.; Cinalli, G. Neurofibromatosis type 1–related hydrocephalus: Causes and treatment considerations. Child’s Nerv. Syst. 2020, 36, 2385–2390.
- Sill Kang, Y.; Park, E.; Kim, Y.; Kim, J.; Kim, D.; Thomale, U.W.; Shim, K.W. Altered cerebrospinal fluid dynamics in neurofibromatosis type l: Severe arachnoid thickening in patients with neurofibromatosis type 1 may cause abnormal CSF dynamic. Childs Nerv. Syst. 2017, 33, 767–775.
- Tanrikulu, B.; Özek, M.M. Neurofibromatosis and Hydrocephalus. In Pediatric Hydrocephalus; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1107–1118.
- Glombova, M.; Petrak, B.; Lisy, J.; Zamecnik, J.; Sumerauer, D.; Liby, P. Brain gliomas, hydrocephalus and idiopathic aqueduct stenosis in children with neurofibromatosis type 1. Brain Dev. 2019, 41, 678–690.
- Roth, J.; Ber, R.; Wisoff, J.H.; Hidalgo, E.T.; Limbrick, D.D.; Berger, D.S.; Thomale, U.W.; Schulz, M.; Cinalli, G.; Santoro, C.; et al. Endoscopic Third Ventriculostomy in Patients with Neurofibromatosis Type 1: A Multicenter International Experience. World Neurosurg. 2017, 107, 623–629.
- Kaas, B.; Huisman, T.A.G.M.; Tekes, A.; Bergner, A.; Blakeley, J.O.; Jordan, L.C. Spectrum and Prevalence of Vasculopathy in Pediatric Neurofibromatosis Type 1. J. Child. Neurol. 2012, 28, 561–569.
- Rerat, K.; Parker, F.; Nasser, G.; Vidaud, D.; Riant, F.; Tournier-Lasserve, E.; Denier, C. Occurrence of multiple Cerebral Cavernous Malformations in a patient with Neurofibromatosis type 1. J. Neurol. Sci. 2015, 350, 98–100.
- Schievink, W.I.; Riedinger, M.; Maya, M.M. Frequency of incidental intracranial aneurysms in neurofibromatosis type 1. Am. J. Med. Genet. Part A 2005, 134A, 45–48.
- Morvan, T.; De Broucker, F.; De Broucker, T. Subarachnoid hemorrhage in neurofibromatosis type 1: Case report of extracranial cerebral aneurysm rupture into a meningocele. J. Neuroradiol. 2011, 38, 125–128.
- Cairns, A.G.; North, K.N. Cerebrovascular dysplasia in neurofibromatosis type 1. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1165–1170.
- Barreto-Duarte, B.; Andrade-Gomes, F.H.; Arriaga, M.B.; Araujo-Pereira, M.; Cubillos-Angulo, J.M.; Bruno Andrade, B. Association between neurofibromatosis type 1 and cerebrovascular diseases in children: A systematic review. PLoS ONE 2021, 16, e0241096.
- Santoro, C.; Giugliano, T.; Kraemer, M.; Torella, A.; Schwitalla, J.C.; Cirillo, M.; Melis, D.; Berlit, P.; Nigro, V.; Perrotta, S.; et al. Whole exome sequencing identifies MRVI1 as a susceptibility gene for moyamoya syndrome in neurofibromatosis type 1. PLoS ONE 2018, 13, e0200446.
- Brandicourt, P.; Bonnet, L.; Béjot, Y.; Drouet, C.; Moulin, T.; Thines, L. Moya-Moya syndrome after cranial radiation for optic glioma with NF1. Case report and literature review of syndromic cases. Neurochirurgie 2018, 64, 63–67.
- Rea, D.; Brandsema, J.F.; Armstrong, D.; Parkin, P.C.; DeVeber, G.; MacGregor, D.; Logan, W.J.; Askalan, R. Cerebral Arteriopathy in Children With Neurofibromatosis Type 1. Pediatrics 2009, 124, e476–e483.
- Paschoal, J.K.S.F.; Paschoal, F., Jr.; de Lima, F.; Pinho, R.; Pereira Vilanova, L.; Bor-Seng-Shu, E.; Masruha, M.R. Detection of Cerebral Vasculopathy by Transcranial Doppler in Children With Neurofibromatosis Type 1. J. Child. Neurol. 2016, 31, 351–356.
- D’Amico, A.; Ugga, L.; Cocozza, S.; Giorgio, S.M.D.A.; Cicala, D.; Santoro, C.; Melis, D.; Cinalli, G.; Brunetti, A.; Pappatà, S. Multimodal evaluation of the cerebrovascular reserve in Neurofibromatosis type 1 patients with Moyamoya syndrome. Neurol. Sci. 2021, 42, 655–663.

