 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vasiliki Sarli | + 3042 word(s) | 3042 | 2021-04-15 08:41:39 | | | |
| 2 | Vivi Li | Meta information modification | 3042 | 2021-04-20 07:32:07 | | |
Video Upload Options
The fusion of 1,2,4-triazole and 1,3,4-thiadiazole rings results in a class of heterocycles compounds with an extensive range of pharmacological properties. A series of 1,2,4-triazolo[3,4-b]-1,2,4-thiadiazoles was synthesized and tested for its enzyme inhibition potential and anticancer activity. The results show that 1,2,4-triazolo[3,4-b]-1,2,4-thiadiazoles display potent anticancer properties in vitro against a panel of cancer cells and in vivo efficacy in HT-29 human colon tumor xenograft in CB17 severe combined immunodeficient (SCID) mice.
1. Introduction
Akt, termed protein kinase B (PKB), is a serine/threonine kinase composed of three isoforms: Akt1, Akt2, and Akt3. All three isoforms share very similar amino acid sequences, with their expression level differentiated; Akt1 and Akt2 are abundantly expressed, while Akt3 is particularly detected in the brain, heart, and kidneys [1][2]. All Akt isoforms display the same basic structure comprised of three regions: (1) an amino terminal pleckstrin homology (PH) domain that interacts with membrane phospholipids such as phosphatidylinositol-3,4,5-triphosphate (PIP3) and phosphatidylinositol 4,5-bisphosphate (PIP2); (2) a central kinase domain that contains the threonine regulatory residue, Thr308, in which phosphorylation activates Akt; and (3) a carboxyl-terminal regulatory domain that consists of a hydrophobic region of 40 amino acids including the serine regulatory residue (Ser473) [3]. The structural elements, PH domain, and the regulatory residues Thr308 and Ser473 play critical roles in the activation of Akt. Two events are required in Akt’s activation: (a) PH-domain-dependent translocation to the plasma membrane and (b) phosphorylation at the Thr308 and Ser473 residues. The first step includes the interaction of the PH domain with PIP3, which is followed by the translocation of Akt to the plasma membrane. Afterwards, Akt adopts a new conformation such that the Thr308 residue would be phosphorylated by phosphoinositide-dependent kinase-1 (PDK1). This signaling event leads to phosphorylated Ser473 via the mechanistic targeting of rapamycin (mTOR)C2 [4][5][6].
Phosphatidylinositol 3-kinase (PI3K), an upstream signaling molecule, along with Akt constitute the PI3K/Akt signaling transduction pathway through which cellular survival and growth are induced in response to extracellular signal [7]. Among all protein components of the PI3K/Akt pathway, the inhibition of Akt has been widely explored due to its association with tumor progression and aggressiveness [8]. Significant alterations have been demonstrated in the expression levels of Akt isoforms in certain malignancies, for instance, Akt1 is particularly elevated in breast, prostate, and gastric tumors whilst Akt2 is overexpressed in prostate, ovarian, breast, pancreatic, and colorectal cancers [9][10][11]. Thus far, Akt inhibitors are divided in four basic categories: (1) ATP-competitive inhibitors, (2) allosteric inhibitors, (3) lipid-based inhibitors, and 4) PH domain inhibitors [12]. The first category of ATP-competitive inhibitors (CCT128930 as an Akt2 inhibitor and BAY-1125976 as an Akt1/2 inhibitor), including pan-Akt kinase inhibitors (afuresertib, GSK690693, AZD5363, GDC-0068, and AT7867 as inhibitors of all Akt isoforms), targets the kinase domain and precisely binds to the ATP-binding pocket [13]. The high degree of homology of the ATP-binding site among different serine/threonine kinases as well as the extensive conservation of this domain within the AGC kinase family (protein kinase A, G, and C families) contributes to a low specificity of ATP-competitive inhibitors. The development of such molecules can be further obstructed due to the lack of strong efficacy against tumors in vivo, with toxicities to normal tissues being recorded at the same time [14][15]. Regarding allosteric inhibitors, for example, the MK-2206 compound, are associated with the Akt kinase domain. Compared with ATP-competitive inhibitors that show efficacy only against cancer cell lines with Akt mutations, allosteric inhibitors display broader anticancer activity as compounds of this type are potent against cancer cell lines with PI3KCA mutations or loss of phosphatase and tensin homologue (PTEN) activity [16]. The third category of lipid-based inhibitors (PX-866 and perifosine) blocks the interaction of Akt with PIP3 since this type of molecule inhibits PI3K and therefore prevents the production of PIP3 from PIP2. Finally, PH domain inhibitors (triciribine and PX-316) inactivate Akt via interactions with the PH-domain, interrupting membrane translocation, which is required for activating Akt [17].
The fusion of 1,2,4-triazole and 1,3,4-thiadiazole rings results in a class of heterocyclic compounds with an extensive range of pharmacological properties including antifungal, antibacterial, antiviral, anti-inflammatory, analgesic, and anthelmintic properties [18]. Earlier studies indicated that previous 3,6-disubstituted 1,2,4-[3,4-b]thiadiazoles induced potent anti-inflammatory activity along with a minimal ulcerogenic effect and lipid peroxidation compared to ibuprofen and flurbiprofen. Some of these compounds demonstrated moderate to weak antibacterial activity against Staphylococcus aureus and Escherichia coli while their antifungal activity against Candida albicans was quite weak [19][20][21]. Furthermore, 1,2,4-triazolo[3,4-b][1,3,4]thiadiazole derivatives containing 3-methyl or benzyl moiety showed moderate anti-HIV-1 activity at subcytotoxic concentrations [22]. In previous work, we have shown that triazolo[3,4-b]thiadiazole derivatives show potent in vitro antiproliferative activities [23]. Our studies resulted in the identification of three bioactive compounds (KA39, KA25, and KA26) against three human colorectal cancer cell lines (Figure 1). Among them, KA39 was the most potent anticancer agent and inhibitor of topIIα phosphorylation at Ser-1106 as well. Additional molecular docking studies revealed that KA39 can occupy the same binding site as etoposide and can interact with the ATPase domain of topIIα, but the exact mechanism of inhibition of topIIα phosphorylation by KA39 is still unclear. As part of our ongoing work on the assessment of the biological activity of triazolo[3,4-b][1,3,4]thiadiazoles, we report the synthesis, structural characterization, and evaluation of inhibitory effects of fifteen new derivatives. In particular, we examined the introduction of ethyl or propyl substituents on sulfonamide combined with different substituents on C-6 of the triazolo[3,4-b][1,3,4]thiadiazole template. In the current work, the anticancer activities of all derivatives were primarily assessed in vitro while the most active molecules KA39, KA25, and KA26, according to in vitro screening, were tested in vivo. Further studies were carried out to investigate whether the most potent antitumor compound blocks the phosphorylation of Αkt1 and Αkt2 kinases. The molecular modeling studies indicated that 1,2,4-triazolo[3,4-b]-1,2,4-thiadiazoles bind well to the ATP binding site in Akt1 and Akt2.

Figure 1. Structures of active triazolo[3,4-b][1,3,4]thiadiazoles and overview of their activity.
2. Synthesis of triazolo[3,4-b]thiadiazole Derivatives
2.1. Synthesis of 3,6-disubstituted 1,2,4-triazolo-[3,4-b]-[1,3,4]-thiadiazoles
Synthesis of the 1,2,4-triazolo-[3,4-b]-[1,3,4]-thiadiazoles was accomplished according to the synthetic methodology developed by our group (Scheme 1 and Scheme 2) [23]. After a reaction with oxalyl chloride, 2-(3,4-dimethoxyphenyl)acetic acid 1 was esterified and the corresponding methyl ester reacted with chlorosulfonic acid in CHCl3 to give methyl 2-(2-(chlorosulfonyl)-4,5-dimethoxyphenyl)acetate 2 in 88% yield. Subsequently, sulfonyl chloride 2 furnished the corresponding sulfonamides 3a,b after reaction with Et2NH or iPr2NH. Furthermore, these compounds reacted with hydrazine hydrate to give hydrazides 4a,b that reacted with KOH and CS2 in EtOH to give the potassium thiocarbamates. The latter products were cyclized in the presence of hydrazine hydrate to triazoles 5a,b [24]. The desired 1,2,4-triazolo[3,4-b]-[1,2,4]-thiadiazoles (6a–e, 7a–d, 9a–d, and 10a,b) were finally prepared via the reaction of triazoles 5a,b with POCl3 and various benzoic or cinnamic acids.
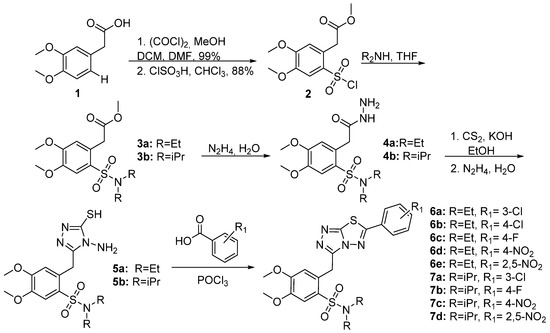
Scheme 1. Synthesis of benzoic acid derivatives.
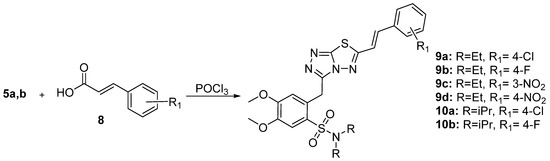
Scheme 2. Synthesis of cinnamic acid derivatives.
2.2. In Vitro Anticancer Activity
The fifteen novel triazolo-thiadiazole derivatives and the previously described KA25, KA26, and KA39 were tested in vitro in two well-established human cancer cell lines: PC-3 and SKOV-3 cells. According to the first drug screening results, the 13 derivatives were inactive at the tested concentrations while the most active compounds (KA25, KA26, KA39, 6e, and 7d) were further tested in vitro in colorectal, ovarian, and prostate cancer cell lines. As Table 1, Table 2 and Table 3 show, the most potent anticancer activities in all human cancer cell lines were induced by the KA39, 6e, and 7d compounds (p < 0.001, two tailed paired t-test). All these compounds bear the 2,5-dinitrophenyl substituent on C-6 of the triazolo[3,4-b][1,3,4]thiadiazole core. Nevertheless, the triazolo[3,4-b]thiadiazole KA39 was significantly more active than 6e and 7d, which displayed less anticancer potency (Table 1, Table 2 and Table 3). Concerning KA25 and KA26 derivatives, both exhibited cytostatic activity in all human cancer cell lines, whereas KA25 induced cytotoxic effects only in ovarian and prostate cancer cells.
Table 1. Growth inhibition/cytostatic (GI50 and TGI) and cytocidal/cytotoxic (IC50) effects induced by triazolo[3,4-b]thiadiazole derivatives KA25, KA26, and KA39 on nine human cancer cell lines. GI50: 50% growth inhibition; TGI: Total growth inhibition; IC50: the concentration that causes 50% cell death.
| Cell Line | KA25 | KA26 | KA39 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| GI50 (μΜ) |
TGI (μΜ) |
IC50 (μΜ) |
GI50 (μΜ) |
TGI (μΜ) |
IC50 (μΜ) |
GI50 (μΜ) |
TGI (μΜ) |
IC50 (μΜ) |
|
| SKOV-3 | 15 ± 0.5 | 50 ± 1.5 | 85 ± 2.1 | 20 ± 0.5 | >100 | >100 | 7 ± 0.2 | 12 ± 0.8 | 25 ± 1.4 |
| UWB 1.289 | 2 ± 0.5 | 5 ± 0.7 | 62 ± 1.2 | 29 ± 0.7 | 53 ± 1 | 70 ± 1.4 | 6 ± 0.2 | 10 ± 0.8 | 22 ± 1.2 |
| UWB1.289+BRCA1 | 4.0 ± 0.5 | 13 ± 0.8 | 38 ± 1.1 | 42 ± 0.5 | 65 ± 0.7 | >100 | 6 ± 0.5 | 11 ± 0.7 | 94 ± 2.3 |
| HT-29 | 1 ± 0.3 | >100 | >100 | 2 ± 0.76 | >100 | >100 | 11.5 ± 0.8 | 15.9 ± 0.55 | 19.5 ± 0.9 |
| LS174T | 9.3 ± 2.0 | 100 ± 0.1 | >100 | 8 ± 1.5 | >100 | >100 | 12 ± 1.5 | 17 ± 1.3 | 21.5 ± 1.5 |
| SW403 | 4.8 ± 0.8 | >100 | >100 | 11 ± 0.8 | >100 | >100 | 5.2 ± 0.2 | 7.9 ± 0.7 | 10 ± 0.76 |
| LoVo | 8 ± 0.76 | 13.8 ± 0.52 | >100 | 15 ± 0.8 | >100 | >100 | 2.2 ± 0.2 | 5.5 ± 0.1 | 10.5 ± 0.15 |
| PC-3 | 14 ± 1.0 | 22 ± 0.8 | >100 | 42.5 ± 1.5 | >100 | >100 | 5 ± 0.15 | 8 ± 0.1 | 12 ± 0.1 |
| DU-145 | 12.8 ± 1.3 | 13.2 ± 0.5 | 27 ± 0.3 | 7 ± 1.0 | 70 ± 1.3 | >100 | 5.8 ± 0.2 | 8 ± 0.4 | 10.3 ± 1.8 |
Table 2. Growth inhibition/cytostatic (GI50 and TGI) and cytocidal/cytotoxic (IC50) induced by the thirteen newly synthesized triazolo[3,4-b]thiadiazoles on two human cancer cell lines.
| Compound | PC-3 | SKOV-3 | ||||
|---|---|---|---|---|---|---|
| GI50 (μΜ) |
TGI (μΜ) |
IC50 (μΜ) |
GI50 (μΜ) |
TGI (μΜ) |
IC50 (μΜ) |
|
| 9b | 42 ± 1.0 | >100 | >100 | >100 | >100 | >100 |
| 10b | 31 ± 1.1 | >100 | >100 | 50 ± 2.0 | >100 | >100 |
| 9a | 11 ± 1.0 | >100 | >100 | 84 ± 2.0 | >100 | >100 |
| 10a | 2 ± 0.5 | 84 ± 2.0 | >100 | 76 ± 6.0 | >100 | >100 |
| 6b | 32 ± 0.4 | 56 ± 0.6 | 104 ± 0.8 | 36 ± 0.1 | 60 ± 0.8 | >100 |
| 6a | 25 ± 1.4 | 112 ± 2.5 | >100 | 6 ± 0.3 | 14 ± 2.5 | >100 |
| 7a | 20 ± 0.8 | >100 | >100 | 16 ± 0.8 | 46 ± 1.5 | >100 |
| 6c | 90 ± 2.0 | >100 | >100 | >100 | >100 | >100 |
| 7b | 25 ± 0.5 | 80 ± 1 | >100 | 80 ± 0.8 | >100 | >100 |
| 6d | 50 ± 0.5 | >100 | >100 | 80 ± 0.5 | >100 | >100 |
| 7c | 23 ± 0.8 | 102 ± 1.2 | >100 | 15 ± 1.4 | 28 ± 2.2 | 66 ± 2.5 |
| 9c | 38 ± 2.0 | >100 | >100 | >100 | >100 | >100 |
| 9d | 30 ± 0.5 | >100 | >100 | >100 | >100 | >100 |
Table 3. Growth inhibition/cytostatic (GI50 and TGI) and cytocidal/cytotoxic (IC50) induced by tri-azolo[3,4-b]thiadiazole derivatives 6e and 7d on five human cancer cell lines.
|
Cell Lines |
6e |
7d |
||||
|---|---|---|---|---|---|---|
|
GI50 (μΜ) |
TGI (μΜ) |
IC50 (μΜ) |
GI50 (μΜ) |
TGI (μΜ) |
IC50 (μΜ) |
|
|
SKOV-3 |
18 ± 0.7 |
24 ± 1.2 |
38 ± 1.8 |
13 ± 0.5 |
17 ± 1.0 |
23 ± 1.4 |
|
PC-3 |
11 ± 1.0 |
17 ± 1.0 |
26.5 ± 0.9 |
8 ± 1.0 |
11 ± 1.04 |
22 ± 0.3 |
|
DU-145 |
13 ± 0.5 |
18.3 ± 0.2 |
28 ± 0.2 |
6.8 ± 0.8 |
14 ± 0.5 |
21.3 ± 0.9 |
|
DLD-1 |
25 ± 2.0 |
37 ± 2.0 |
53.6 ± 1.05 |
7.3 ± 0.75 |
12 ± 0.5 |
22 ± 1.0 |
|
HT-29 |
18 ± 1.0 |
29 ± 0.5 |
44 ± 1.0 |
17.9 ± 2.51 |
26 ± 1.52 |
40 ± 1.0 |
3. Discussion
Aberrations in the PI3K/Akt signal transduction pathway are a very common phenomenon in human carcinogenesis, with over half of tumors showing irregular Akt activation [25]. More precisely, the overexpression of certain oncogenes or a lack of tumor suppressor genes influences the biological activity of the PI3K/Akt network, stimulating cancer cell growth. For instance, mutations of Epidermal Growth Factor Receptor, EGFR/PI3K, loss of the tumor suppressor protein phosphatase and tensin homologue (PTEN), and mutations or amplifications of Akt itself enhance Akt’s signaling in cancer cells [26][27]. Amplified and mutated Akt isoforms play crucial roles in tumorigenesis, while Akt2 is particularly involved in EMT transition and metastasis. Regarding mutations, Akt1 is the most frequently mutated isoform compared with Akt2, in which mutations do not occur at high frequency and cannot be considered as activating [28][29]. The most prevalent mutation in Akt1, E17K, is located within the PH domain and is found in several solid tumors including breast (5.9%), colorectal (1.6%), lung (0.6%), and ovarian cancers (0.8%) as well as in melanoma (0.5%) [30][31]. Akt1E17K mutation, induced by the substitution of lysine to glutamic acid at amino acid 17, leads to a pathological translocation of Akt1 to the plasma membrane and to alterations in PIP specificity as binding to PIP2 seems to be highly increased. Given that Akt1 normally binds to PIP3, further utilization of PIP2 in the case of Akt1E17K enhances Akt1′s signaling [31][32][33].
Tumor aggressiveness as well as poor survival rates are particularly linked to Akt2′s amplification and overexpression [9]. According to earlier studies, mutations in the Akt2 isoform are quite rare whilst amplifications in the isoform have been detected in 16% of pancreatic cancers; 16% of uterine cancers; 13% of breast cancers; and 5–10% of ovarian, lung, and bladder cancers. The role of Akt2 in oncogenesis is reflected by a reduction in cell growth and invasiveness induced by siRNA depletion of Akt2 in cells that overexpress Akt. On the other hand, an amplified Akt1 is less frequent since it has been found only in 20% of neuroendocrine prostate cancers, 10% of pancreatic cancers, and 3–5% of breast and serous ovarian cancers [34]. Altogether, Akt is strongly implicated in many types of cancer and therefore has been validated as a therapeutic target nearly two decades ago [35].
The present study attempts to explore the inhibitory impact of KA25 and KA39 derivatives on Akt1 and Akt2 phosphorylation in three colorectal cancer cell lines (HT-29, LoVo, and SW403) with specific PIK3CA and KRAS statuses (Table 4). More specifically, two of the referred cell lines, HT-29 and SW403, harbor mutations in PIK3CA, p.P449T and p.Q546K, respectively, while LoVo cells retain a wild-type PIK3CA (https://cancer.sanger.ac.uk/cosmic, accessed on 4 September 2020). With regard to PIK3CA mutations, most of them are concentrated in hotspots within two domains: the helical (exon 9) and kinase (exon 20) [36]. Nonetheless, four additional missense mutations have been detected including p.P449T, which belongs to gain-of-function mutations and contributes to enhanced PIK3CA kinase activity (>2-fold) compared to the wild-type PIK3CA. In general terms, gain-of-function mutations in PIK3CA are known for triggering the PI3K kinase activity and consequently activating its downstream molecules such as Akt [37]. Further studies have indicated that rare PIK3CA mutants, including p.Q546K, possess a high oncogenic potency as well as hyperactivate PI3K/Akt downstream signaling cascade [38]. Due to the fact that HT-29 and SW403 cell lines contain gain-of-function mutations in PIK3CA, the levels of phosphorylated Akt may be elevated in both cell lines in comparison to the wild-type PIK3CA expressed by the LoVo cell line.
Table 4. Description of histotypes and special characteristics of the three human colorectal cancer cell lines in which the inhibition of Akt (Akt1/2) phosphorylation, induced by KA25 and KA39, was investigated.
| Cancer Type | Human Cell Line Designation | KRAS Status | PIK3CA Status | References |
|---|---|---|---|---|
| Colorectal adenocarcinoma | HT-29 | wild-type | p.P449T | [39][40] |
| Colorectal adenocarcinoma, Duke’s type C, grade IV | LoVo | p.G13D | wild-type | [39][40] |
| Colorectal adenocarcinoma Dukes’ type C, grade III | SW403 | p.G12V | p.Q546K | [41][42][43] |
With respect to KRAS, two of the three cell lines, LoVo and SW403, are KRAS-mutant-bearing p.G13D and p.G12V mutations, respectively (Table 6). Experimental studies using these cell lines showed that neither p.G12V nor p.G13D KRAS mutations stimulate the phosphorylation of Akt to a greater extent than wild-type KRAS cells. Moreover, it has been also demonstrated that Akt phosphorylation was decreased in KRAS-G12V cells compared to KRAS wild-type cell lines [44][45][46]. Consequently, the LoVo and SW403 cell lines (KRAS-mutant) probably express similar or lower levels of phosphorylated Akt than HT-29 cells (KRAS wild-type). As far as cell sensitivity to Akt inhibitors is concerned, recent studies support that cell lines with PI3K and/or PTEN mutations display a higher susceptibility to this type of inhibitor than cells with KRAS and/or v-raf murine sarcoma viral oncogene homolog B (BRAF) mutations [47]. More specifically, Akt inhibitors show greater selectivity and potency in cells with increased Akt kinase activity resulting from mutations in PI3K or PTEN. Later studies implied that cell sensitivity to allosteric Akt1/2 inhibitors is significantly associated with Akt phosphorylation in human cancer cell lines, leading to the conclusion that cell lines with elevated levels of phosphorylated Akt are more susceptible to the pharmacologic inhibition of Akt1/2. However, the cell lineage may affect the pharmacologic sensitivity to Akt1/2 inhibition, for instance, PIK3CA-mutant breast cancer cell lines are more sensitive than PIK3CA-mutant colon cancer cell lines. Altogether, high levels of phosphorylated Akt signify AKT dependency in PIK3CA-mutant cancer cells [48][49].
On the other hand, Akt inhibitors, utilized in the inhibition of downstream effector pathways, have not shown an impressive antitumor efficacy on KRAS-mutant tumors [50]. As observed, cells with mutated KRAS and/or BRAF are less sensitive to Akt inhibitors since no alterations in the activation of MAPK pathway have arisen [47]. A relevant example that correlates PIK3CA and RAS mutations with sensitivity to Akt inhibitors concerns the AZD5363 compound; PIK3CA and PTEN mutations are strongly associated with sensitivity to this Akt inhibitor, regardless of RAS status [51]. Additionally, tumors bearing an overactivated PI3K/Akt pathway without any EGFR, RAS, or BRAF mutations depend more on this pathway and become more sensitive to selective Akt inhibitors [52]. It is noteworthy that both of the referred to KRAS mutations (p.G12V and p.G13D) are associated with resistance to anti-EGFR therapy via intracellular pathways, restricting the administration of anti-EGFR monoclonal antibodies (mAbs) to patients bearing KRAS wild-type tumors [53][54]. Based on PIK3CA and KRAS statuses, we could put forward the hypothesis that KA25- and KA39-induced inhibition of Akt1 and Akt2 phosphorylation might be more effective in HT-29 (PIK3CA-mutant; KRAS wild-type) than those of LoVo cells (PIK3CA wild-type; KRAS-mutant). Regarding SW403 cells, in which PIK3CA and KRAS mutations concur, Akt1 and Akt2 phosphorylation could be impeded similarly to either HT-29 or LoVo cells. Nevertheless, both methods of estimation (% inhibition and ratios correlated with control) indicate that both triazolo[3,4-b]thiadiazole derivatives exhibited significant inhibitory activity against Akt1and Akt2 phosphorylation in all three cancer cell lines. It is notable that the inhibition of Akt1 and Akt2 phosphorylation, induced by KA25 and KA39, was fairly similar in all three cell lines carrying PIK3CA and/or KRAS mutations. Nonetheless, it could be suggested that the reduction in phosphorylated Akt (Akt1/2) was slightly greater in LoVo (mutated KRAS) and SW403 (mutated KRAS and PIK3CA) cells compared to the HT-29 cell line (mutated PIK3CA).
Furthermore, pharmacokinetic studies on the tested compounds are currently under investigation. Premature experimental data showed that these compounds have poor water solubility (log mol/L = −4.4 to −3.5), high CaCO2 permeability (log Papp in 10−6 cm/s = −0.135 to 0.965), and high intestinal absorption (94–100%). These compounds present low distribution volumes with high binding to serum proteins (72–92%), are unable to penetrate the CNS, and are poorly distributed to the brain. They are not substrates of P-glycoprotein but they inhibit both glycoproteins I and II. They metabolize in liver, and they are substrates of CYP3A4 but inhibit CYP2C19 and CYP2C19. Their total clearance in rats was 2.2–5.7 mL/min/kg.
It is interesting to note that a large number of small molecules are proven to inhibit Akt in vitro and in vivo, though only a limited number of these have entered clinical trials [40]. Ongoing clinical studies indicate that the tested Akt inhibitors are not effective as single agents, displaying rather low therapeutic responses. Furthermore, even if there is increasing knowledge concerning Akt functions and activation, there are no Akt inhibitors approved for oncologic use. For example, miltefosine, the only Akt inhibitor approved, is intended for treating visceral and cutaneous leishmaniasis and not cancer disease [30][40]. Therefore, the discovery of drugs such as KA39 constitutes a valuable tool in the research area of Akt inhibitors due to its high anticancer potency and multitarget behavior.
References
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927.
- Martelli, A.M.; Tabellini, G.; Bressanin, D.; Ognibene, A.; Goto, K.; Cocco, L.; Evangelisti, C. The emerging multiple roles of nuclear Akt. Biochim. Biophys. Acta 2012, 1823, 2168–2178.
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell Mol. Med. 2005, 9, 59–71.
- Andjelković, M.; Alessi, D.R.; Meier, R.; Fernandez, A.; Lamb, N.J.; Frech, M.; Cron, P.; Cohen, P.; Lucocq, J.M.; Hemmings, B.A. Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 1997, 272, 31515–31524.
- Carnero, A.; Paramio, J.M. The PTEN/PI3K/AKT Pathway in vivo Cancer Mouse Models. Front Oncol. 2014, 4, 252.
- Conus, N.M.; Hannan, K.M.; Cristiano, B.E.; Hemmings, B.A.; Pearson, R.B. Direct identification of tyrosine 474 as a regulatory phosphorylation site for the Akt protein kinase. J. Biol. Chem. 2002, 277, 38021–38028.
- Arcaro, A.; Guerreiro, A.S. The phosphoinositide 3-kinase pathway in human cancer: Genetic alterations and therapeutic implications. Curr. Genom. 2007, 8, 271–306.
- Mitsiades, C.S.; Mitsiades, N.; Koutsilieris, M. The Akt pathway: Molecular targets for anti-cancer drug development. Curr. Cancer Drug Targets 2004, 4, 235–256.
- Rychahou, P.G.; Kang, J.; Gulhati, P.; Doan, H.Q.; Chen, L.A.; Xiao, S.Y.; Chung, D.H.; Evers, B.M. Akt2 overexpression plays a critical role in the establishment of colorectal cancer metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 20315–20320.
- Graff, J.R.; Konicek, B.W.; McNulty, A.M.; Wang, Z.; Houck, K.; Allen, S.; Paul, J.D.; Hbaiu, A.; Goode, R.G.; Sandusky, G.E.; et al. Increased AKT activity contributes to prostate cancer progression by dramatically accelerating prostate tumor growth and diminishing p27Kip1 expression. J. Biol. Chem. 2000, 275, 24500–24505.
- Altomare, D.A.; Tanno, S.; De Rienzo, A.; Klein-Szanto, A.J.; Tanno, S.; Skele, K.L.; Hoffman, J.P.; Testa, J.R. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J Cell Biochem. 2002, 87, 470–476.
- Pal, S.K.; Reckamp, K.; Yu, H.; Figlin, R.A. AKT inhibitors in clinical development for the treatment of cancer. Expert Opi. Investig. Drugs 2010, 19, 1355–1366.
- Brown, J.S.; Banerji, U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017, 172, 101–115.
- Yang, J.; Cron, P.; Thompson, V.; Good, V.M.; Hess, D.; Hemmings, B.A.; Barford, D. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif 587 phosphorylation. Mol. Cell. 2002, 9, 1227–1240.
- Hu, Y.; Qiao, L.; Wang, S.; Rong, S.B.; Meuillet, E.J.; Berggren, M.; Gallegos, A.; Powis, G.; Kozikowski, A.P. 3-(Hydroxymethyl)-bearing phosphatidylinositol ether lipid analogues and carbonate surrogates block PI3-K, Akt, and cancer cell growth. J. Med. Chem. 2000, 43, 3045–3051.
- Lindsley, C.W.; Barnett, S.F.; Yaroschak, M.; Bilodeau, M.T.; Layton, M.E. Recent progress in the development of ATP-competitive and allosteric AKT kinase inhibitors. Curr. Top. Med. Chem. 2007, 7, 1349–1363.
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274.
- Charitou, T.; Srihari, S.; Lynn, M.; Jarboui, M.-A.; Fasterius, E.; Moldovan, M.; Shirasawa, S.; Tsunoda, T.; Ueffing, M.; Xie, J.; et al. Transcriptional and metabolic rewiring of colorectal cancer cells expressing the oncogenic KRAS G13D mutation. Br. J. Cancer 2019, 121, 37–50.
- Amir, M.; Kumar, H.; Javed, S.A. Condensed bridgehead nitrogen heterocyclic system: Synthesis and pharmacological activities of 1,2,4-triazolo-[3,4-b]-1,3,4-thiadiazole derivatives of ibuprofen and biphenyl-4-yloxy acetic acid. Eur. J. Med. Chem. 2008, 43, 2056–2066.
- Karabasanagouda, T.; Adhikari, A.V.; Shetty, N.S. Synthesis and antimicrobial activities of some novel 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles and 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazines carrying thioalkyl and sulphonyl phenoxy moieties. Eur. J. Med. Chem. 2007, 42, 521–529.
- Chawla, G.; Kumar, U.; Bawa, S.; Kumar, J. Syntheses and evaluation of anti-inflammatory, analgesic and ulcerogenic activities of 1,3,4-oxadiazole and 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole derivatives. J. Enz. Inhib. Med. Chem. 2012, 27, 658–665.
- Kritsanida, M.; Mouroutsou, A.; Marakos, P.; Pouli, N.; Papakonstantinou-Garoufalias, S.; Pannecouque, C.; Witvrouw, M.; De Clercq, E. Synthesis and antiviral activity evaluation of some new 6-substituted 3-(1-adamantyl)-1,2,4-triazolo[3,4-b][1,3,4]thiadiazoles. Farmaco 2002, 57, 253–257.
- Sagredou, S.; Dalezis, P.; Nikoleousakos, N.; Nikolaou, M.; Voura, M.; Almpanakis, K.; Panayiotidis, M.I.; Sarli, V.; Trafalis, D.T. 3,6-Disubstituted 1,2,4-Triazolo[3,4-b]Thiadiazoles with Anticancer Activity Targeting Topoisomerase II Alpha. Onco. Targets Ther. 2020, 13, 7369–7386.
- Reid, J.R.; Heindel, N.D. Improved syntheses of 5-substituted-4-amino-3-mercapto-(4H)-1,2,4-triazoles. J. Heterocycl. Chem. 1976, 13, 925–926.
- You, I.; Erickson, E.C.; Donovan, K.A.; Eleuteri, N.A.; Fischer, E.S.; Gray, N.S.; Toker, A. Discovery of an AKT Degrader with Prolonged Inhibition of Downstream Signaling. Cell Chem Biol. 2020, 27, 66–73.
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004.
- Rhodes, N.; Heerding, D.A.; Duckett, D.R.; Eberwein, D.J.; Knick, V.B.; Lansing, T.J.; McConnell, R.T.; Gilmer, T.M.; Zhang, S.-J.; Robell, K.; et al. Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res. 2008, 68, 2366–2374.
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404.
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, l1.
- Shoji, K.; Oda, K.; Nakagawa, S.; Hosokawa, S.; Nagae, G.; Uehara, Y.; Sone, K.; Miyamoto, Y.; Hiraike, H.; Hiraike-Wada, O.; et al. The oncogenic mutation in the pleckstrin homology domain of AKT1 in endometrial carcinomas. Br. J. Cancer 2009, 101, 145–148.
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444.
- Landgraf, K.E.; Pilling, C.; Falke, J.J. Molecular mechanism of an oncogenic mutation that alters membrane targeting: Glu17Lys modifies the PIP lipid specificity of the AKT1 PH domain. Biochemistry 2008, 47, 12260–12269.
- Kumar, A.; Purohit, R. Cancer associated E17K mutation causes rapid conformational drift in AKT1 pleckstrin homology (PH) domain. PLoS ONE 2013, 8, e64364.
- Cheng, J.Q.; Ruggeri, B.; Klein, W.M.; Sonoda, G.; Altomare, D.A.; Watson, D.K.; Testa, J.R. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc. Natl. Acad. Sci. USA 1996, 93, 3636–3641.
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.Α.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol. 2016, 48, 869–885.
- Ikenoue, T.; Kanai, F.; Hikiba, Y.; Obata, T.; Tanaka, Y.; Imamura, J.; Ohta, M.; Jazag, A.; Guleng, B.; Tateishi, K.; et al. Functional Analysis of PIK3CA Gene Mutations in Human Colorectal Cancer. Cancer Res. 2005, 65, 4562–4567.
- Tsubaki, M.; Takeda, T.; Noguchi, M.; Jinushi, M.; Seki, S.; Morii, Y.; Shimomura, K.; Imano, M.; Satou, T.; Nishida, S. Overactivation of Akt Contributes to MEK Inhibitor Primary and Acquired Resistance in Colorectal Cancer Cells. Cancers 2019, 11, 1866.
- Gymnopoulos, M.; Elsliger, M.; Vogt, P. Rare cancer-specific mutations in PIK3CA shows gain of function. Proc. Natl. Acad. Sci. USA 2007, 104, 5569–5574.
- Ahmed, D.; Eide, P.; Eilertsen, I.; Danielsen, S.A.; Eknæs, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71.
- Berg, K.; Eide, P.; Eilertsen, I.; Johannessen, B.; Bruun, J.; Danielsen, S.A.; Bjørnslett, M.; Meza-Zepeda, L.A.; Eknæs, M.; Lind, G.E.; et al. Multi-omics of 34 colorectal cancer cell lines—A resource for biomedical studies. Mol. Cancer 2017, 16, 116.
- . Broad Institute Cancer Cell Line Encyclopedia (CCLE). Available online: (accessed on 16 September 2020).
- . COSMIC—Catalogue of Somatic Mutations in Cancer. Available online: (accessed on 16 September 2020).
- . Mutation Overview Page PIK3CA—P.Q546K (Substitution—Missense). Available online: (accessed on 4 September 2020).
- Johnson, C.W.; Lin, Y.-J.; Reid, D.; Parker, J.; Pavolopoulos, S.; Dischinger, P.; Graveel, C.; Aguirre, A.J.; Steensma, M.; Haigis, K.M.; et al. Isoform-Specific Destabilization of the Active Site Reveals a Molecular Mechanism of Intrinsic Activation of KRas G13D. Cell Rep. 2019, 28, 1538–1550.e7.
- Stolze, B.; Reinhart, S.; Bullinger, L.; Fröhling, S.; Scholl, C. Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using humanMCF10A isogenic cell lines. Sci. Rep. 2015, 5, 8535.
- Ihle, N.T.; Byers, L.A.; Kim, E.S.; Saintigny, P.; Lee, J.J.; Blumenschein, G.R.; Tsao, A.; Liu, S.; Larsen, J.E.; Wang, J.; et al. Effect of KRAS oncogene substitutions on protein behavior: Implications for signaling and clinical outcome. J. Natl. Cancer Inst. 2012, 104, 228–239.
- Dumble, M.; Crouthamel, M.-C.; Zhang, S.-Y.; Schaber, M.; Levy, D.; Robell, K.; Liu, Q.; Figueroa, D.J.; Minthorn, E.A.; Seefeld, M.A.; et al. Discovery of Novel AKT Inhibitors with Enhanced Anti-Tumor Effects in Combination with the MEK Inhibitor. PLoS ONE 2014, 9, e100880.
- She, Q.; Chandarlapaty, S.; Ye, Q.; Lobo, J.; Haskell, K.M.; Leander, K.R.; DeFeo-Jones, D.; Huber, H.E.; Rosen, N. Breast Tumor Cells with PI3K Mutation or HER2 Amplification Are Selectively Addicted to Akt Signaling. PLoS ONE 2008, 3, e3065.
- Vasudevan, K.M.; Barbie, D.A.; Davies, M.A.; Rabinovsky, R.; McNear, C.J.; Kim, J.J.; Hennessy, B.T.; Tseng, H.; Pochanard, P.; Kim, S.Y.; et al. AKT-Independent Signaling Downstream of Oncogenic PIK3CA Mutations in Human Cancer. Cancer Cell 2009, 16, 21–32.
- Tolcher, A.W.; Khan, K.; Ong, M.; Banerji, U.; Papadimitrakopoulou, V.; Gandara, D.R.; Patnaik, A.; Baird, R.D.; Olmos, D.; Garrett, C.R.; et al. Antitumor activity in RAS-driven tumors by blocking AKT and MEK. Clin. Cancer Res. 2015, 21, 739–748.
- Davies, B.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.-H.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical Pharmacology of AZD5363, an Inhibitor of AKT: Pharmacodynamics, Antitumor Activity, and Correlation of Monotherapy Activity with Genetic Background. Mol. Cancer Ther. 2012, 11, 873–887.
- She, Q.; Halilovic, E.; Ye, Q.; Zhen, W.; Shirasawa, S.; Sasazuki, T.; Solit, D.; Rosen, N. 4E-BP1 Is a Key Effector of the Oncogenic Activation of the AKT and ERK Signaling Pathways that Integrates Their Function in Tumors. Cancer Cell 2010, 18, 39–51.
- Gamba, S.; Camaj, P.; Heinemann, V.; Laubender, R.P.; Wang, Y.; Zhao, Y.; Stintzing, S.; Giessen, C.; Boeck, S.; Haertl, C.; et al. Effect of KRAS exon 2 mutations on antitumor activity of afatinib and gefitinib. Anticancer Drugs 2015, 26, 371–378.
- Song, Q.; Sun, X.; Guo, H.; Yu, Q. Concomitant inhibition of receptor tyrosine kinases and downstream AKT synergistically inhibited growth of KRAS/BRAF mutant colorectal cancer cells. Oncotarget 2017, 8, 5003–5015.


