 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alfredo Berzal-Herranz | + 4350 word(s) | 4350 | 2021-04-06 10:46:22 | | | |
| 2 | Rita Xu | Meta information modification | 4350 | 2021-04-20 10:05:09 | | |
Video Upload Options
The flavivirus genome consists of a single positive-stranded RNA molecule with just one open reading frame (ORF) flanked by untranslated 5′ and 3′ regions. The ORF encodes a polyprotein that is processed to produce three structural and seven non-structural viral proteins. The RNA genome is endowed with a type I cap structure at the 5′ terminus and lacks a poly A tail at its 3′ end. To store all the information required for their successful propagation, flaviviruses use discrete structural genomic RNA elements to code for functional information by the establishment of dynamic networks of long-range RNA–RNA interactions that promote specific functional folding.
1. Introduction
The genus Flavivirus comprises many small, enveloped viruses commonly known as flaviviruses. Of global health importance, they are responsible for many emerging and re-emerging outbreaks of human disease, with dengue virus (DENV), zika virus (ZIKV), Japanese encephalitis virus (JEV), yellow fever virus (YFV) and West Nile virus (WNV) among the most important. Flaviviruses can be largely classified according to their arthropod vector as either mosquito-borne flaviviruses (MBFV), tick-borne flaviviruses (TBFV), no-known vector flaviviruses (NKFV), insect-specific flaviviruses (ISFV), or as the unclassified Tamana bat viruses (TABV) [1][2].
The flavivirus genome consists of a single positive-stranded RNA molecule with just one open reading frame (ORF) flanked by untranslated 5′ and 3′ regions (UTRs). The ORF encodes a polyprotein that is processed to produce three structural and seven non-structural viral proteins [3][4]. The RNA genome is endowed with a type I cap structure at the 5′ terminus and lacks a poly A tail at its 3′ end. Like many other RNA viruses, flaviviruses have developed different strategies to expand their capacity to store information, allowing them to code for all the information required for the successful completion of the viral cycle in a genome of just ~11,000 nucleotides. Besides containing protein-coding information, the flavivirus RNA genome stores essential information in structurally conserved units. These are scattered throughout the genome, with the flanking UTRs being rich in them (reviewed by [5][6]). Thus, RNA genomes behave as molecules that store protein-coding information (mRNA), and as non-coding RNAs (ncRNAs), functions that together ensure the correct regulation of the viral cycle. This multifunctional behavior is maintained by the establishment of intricate networks of RNA–RNA interactions—the RNA interactome—that allow the formation of high-order structures critical to infection.
2. Structural Diversification of the Flavivirus RNA Genome
All flaviviruses bear UTRs at both ends of their genome; these are essential for viral viability. Their functioning relies on their structure, which varies widely among members of different ecological groups and even different clades of the same group. UTR folding involves a number of discrete, functionally active structural RNA units with defined roles. It is noteworthy that, regardless of their different folding, overall UTR functioning is preserved across the flaviviruses, revealing that this functionality can be achieved with different RNA conformations.
2.1. 5′ Untranslated Region
The 5′UTR is ~100 nt long [7]. Although its sequence varies somewhat across flaviviruses, the overall RNA structure is largely conserved (with some more notable differences between members of the MBFV and TBFV groups). The 5′UTR folds into two conserved domains (Figure 1): stem-loop A (SLA), which operates as a promoter of viral replication [8][9][10], and stem-loop B (SLB), which contains the essential 5′UAR (upstream AUG region) motif required for genome cyclization (see below) [11][12]. Both domains are linked by a short U-rich sequence, which can pair with the viral translation starting region to generate the 5′UAR-flanking stem (UFS) (Figure 1) [13]. The UFS is present in most MBFV members, in some representatives of the NKFV, and in different members of the ISFV group [13], while the YFV clade and certain members of NKFV show a different folding pattern resulting in a structure termed ψUFS (Figure 1) [13]. The UFS and ψUFS structures of the MBFV and NKFV members contribute to the recruitment of the viral polymerase NS5 while the viral genome is in linear form [13][14]. In the ISFV, its function remains unknown. Intriguingly, this structure is absent in the TBFV; rather, a short hairpin with a large apical loop (5′SL2) is located in an analogous position (Figure 1b) [13]. This suggests that flaviviruses may have developed alternative mechanisms for the control of RNA synthesis [13].
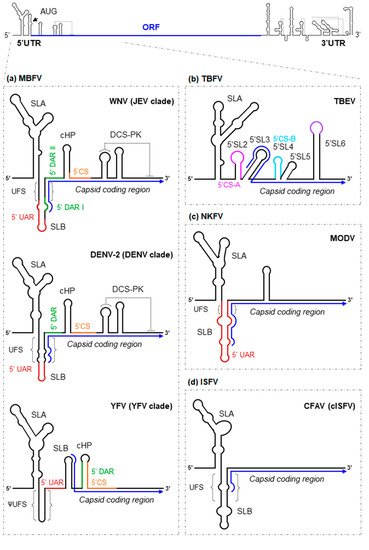
Figure 1. Structural RNA elements of the 5′ end of representative flavivirus genomes. As a model for flaviviruses, a diagram of the WNV genome is shown at the top of the figure. The arrowhead indicates the position of the AUG translation initiation codon. The ORF is represented by a thick blue line. Enlarged are diagrams representing the main functional–structural RNA elements and sequence motifs identified at the 5′ end of the genome of representative members of the: (a) mosquito-borne flaviviruses group (represented by WNV, DENV-2 and YFV); (b) tick-borne flaviviruses group (represented by TBEV); (c) No-known vector flaviviruses group (represented by MODV); and (d) insect-specific flaviviruses group (represented by CFAV). The blue arrow line indicates the ORF. Structural elements and sequence motifs are named. Sequence motifs involved in genome cyclization (UAR, DAR and CS) are indicated by colored lines. Dotted lines indicate interactions involved in the formation of pseudoknot structures.
2.2. 3′ Untranslated Region
The 3′UTR region is between 400 and 800 nt long. A remarkable feature of this region is the existence of structural duplications. The existence of alternative folding intermediates dependent on duplicated elements increases the functional diversity. Indeed, such functional diversification is known to exist, providing, for example, adaptation to different hosts.
The 3′UTR is organized into three major domains in the MBFV (Figure 2) [15]:
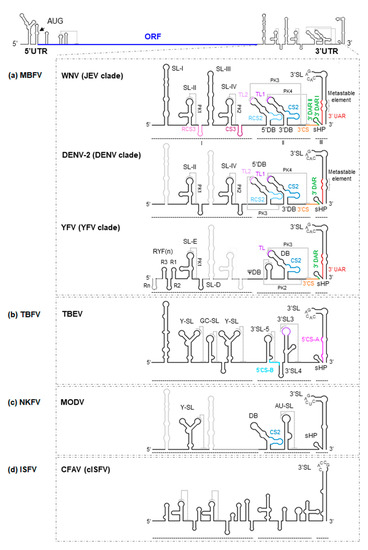
Figure 2. Secondary structure model of the 3′UTR of flavivirus genomes. As a model for flaviviruses, a diagram of the WNV genome is shown at the top of the figure. Below are detailed representations of the secondary structure of the genomic 3′UTR, indicating the identified secondary structural elements, the tertiary interactions and sequence motifs, for (a) WNV, DENV-2 and YFV, all members of the MBFV group; (b) TBEV, representative of the TBFV group; (c) MODV, a member of the NKFV group; and (d) CFAV, a member of the ISFV group. Secondary structural elements of each virus are indicated (thick black lines) on the secondary structure model of the WNV background (thick grey lines); note the differences between them. Structural elements and sequence motifs are named. Sequence motifs involved in genome cyclization (UAR, DAR and CS) are indicated by colored lines (same key as in Figure 1) to designate corresponding sequence partners in both figures. Dotted lines indicate interactions involved in the formation of pseudoknot structures. Thick dotted lines in the representation of YFV secondary structure indicate the presence of extra RYF elements in other members of the YFV clade.
(i) Domain I, located downstream from the translation stop codon, contains a variable structural element followed by a number of conserved stem-loops (SLs). Indeed, significant variations in the number of SLs have been reported. For example, some members of the MBFV group, such as YFV, possess tandem duplications of two SLs (known as RYF) (Figure 2a) [16]. While DENV shows a duplicated SL—SL-I and SL-II (Figure 2a)—other representative MBFV members, such as those belonging to the JEV clade, have duplicated this cassette and thus have four SL units (SL-I to SL-IV) (Figure 2a) [17][18][19].
The existence of SL duplications in domain I of different flaviviruses may be an adaptive mechanism that assists in the movement between natural hosts without interfering with any other activities of the 3′UTR [20]. For example, in DENV, point mutations in SL-II that reduce its structural stability led to a loss of replication efficacy in mammalian cells [21], but not in mosquito cells [22].
An important structural feature of flavivirus RNA genomes is that they bear a variable number of conserved pseudoknot structures (PK) in domain I (Figure 2). They involve the residues located at the apical loop of each SL and the nearby downstream sequences. These SL-PK elements influence infectivity, host adaptation and viral fitness [21][23][24][25], and are likely related to the stalling of cellular exoribonuclease XRN1, leading to the production of sfRNAs [26] (see below). Importantly, when a single SL-PK structural element is borne, as in YFV (PK1; Figure 2a), virulence correlates with the stability of that SL-PK [27].
(ii) Domain II shows moderate variations in its structural organization, not only between different ecological groups but also among different clades of the same group [28]. For example, in members of the JEV and DENV clades within the MBFV group, domain II shows two dumbbell (DB)-like structures, the 5′ and 3′DBs [29], with a linker sequence varying in sequence and length (Figure 2a) [28]. In contrast, YFV bears a single pseudo-DB element, ψDB, the sequence of which seems to be a combination of the duplicated 5′ and 3′DB elements present in other MBFV members (Figure 2a) [17][20][24]. In addition, it bears a complete DB structural element. Representatives of NKFV, such as MODV (Table 1), have a single DB element (Figure 2c), while in ISFV and TBFV, this kind of structure is substituted by moderately conserved SLs (Figure 2b,d) [24][30]. Interestingly, duplicated elements commonly undergo functional specialization, as can be inferred from the sequence analysis of the DB elements in DENV. In this virus, the homology between the 5′ and 3′DB is less than that observed between DB elements of different viral serotypes [20], a consequence of the different selective pressure placed on each DB element in adult mosquitoes. Only the 3′DB element shows sequence variation (it can be even lost), while the 5′DB element is critical to the preservation of efficient replication. These observations support the idea that DB duplications can widen the range of genome functionality, something that might occur in all members of the MBFV group [20]. In MBFV, the DB elements also play a role in translation [31], although their involvement in the generation of sfRNAs and replication are their most significant contributions to the viral cycle [31][32]. Again, duplicated elements provide for different structure-based viral adaptations [21].
A significant feature of flavivirus genomes with double DB elements is the formation of at least one PK structure stabilized by four-way junctions (Figure 2). The PK elements within domain II involve sequences located at the apical loops (named TLs) of the DB and the corresponding downstream complementary motifs. Since sequences interacting with the TLs overlap conserved sequences (CS) required for genomic cyclization (Figure 2) [19][24][33], it is likely that these PK structures compete for elements that are essential in genomic cyclization, preventing the latter from occurring (see below; [20][25][34]). This competition may provide a means of regulating switching between different steps of the viral cycle.
The DB elements also incorporate conserved and reverse-conserved sequences, named CS2 and RCS2, respectively (Figure 2) [29][35]. These sequences of 20 to 45 nt are mostly conserved in flaviviruses [29]. CS2 appears in the DENV, JEV and YFV clades, as well as in the NKFV group, while RCS2 is absent from YFV and NKFV (Figure 2). Duplications of conserved sequences may have evolved to maximize protein recruitment and thus enhance replication, translation, etc., but this remains to be confirmed.
(iii) Domain III is the most structurally conserved region within the 3′UTR. It contains a large terminal SL-3′SL, preceded by a small hairpin structure, sHP (Figure 2). Both 3′SL and sHP are preserved across MBFV, TBFV and NKFV [30][36], while sHP is absent in cISFV [37].
The sHP element is a small hairpin carrying different concatenate sequence motifs (UAR, DAR (downstream AUG region) and CS) and is involved in genomic cyclization. It can adopt different conformations in the cyclized and linear forms [38]. Systematic mutation methods used with DENV have revealed the preservation of the hairpin folding to be essential for the replication of the virus in mammalian cells, though tolerance to nucleotide mutations is relatively high. However, sequence variations can interfere significantly with replication in mosquito cells, suggesting a double function for sHP, both at the sequence level and the structural level [22]. The strong conservation of sHP across all ecological groups confirms this element to be essential to flaviviruses fitness.
3′SL bears a conserved pentanucleotide sequence motif (5′-CACAG-3′) in the apical loop [37][39][40] that is required for replication [41][42][43][44] and the potentiation of translation [45][46]. These functions are achieved via the recruitment of different proteins including viral NS5 polymerase [47], eukaryotic elongation factor eEF-1α [48][49][50], T-cell intracellular antigen-1 (TIA-1) and TIA-1-related (TIAR) proteins [51][52][53], Y box-binding protein-1 (YB-1; [54]), interleukin enhancer binding factor 3 (ILF3, also referred as nuclear factor 90 [NF90] [55]), DEAH box polypeptide 9 (DHX9, also known as RNA helicase A [RHA] [55]), and La protein [56][57].
Two conserved base pairs can be also found at the base of the 3′SL element in the JEV and DENV clades, which are flanked by a metastable structure identified as two symmetrical bulges (Figure 2) [58][59]. This region stabilizes the whole 3′SL structure, and it has been proposed to be involved in the switch from the linear to the cyclized genome [59][60]. Indeed, the 5′ basal segment of the 3′SL contains the 3′UAR motif, which is essential for genome cyclization (Figure 2) ([44][60]; see below). The unwinding of this segment is required for the efficient replication of MBFV in mammalian cells, while in mosquito cells, such structural destabilization occurs only marginally (see below; [59]).
It would thus appear that a dynamic network of RNA–RNA interactions regulates the critical processes of the viral life cycle and the switches between them. The diversification of RNA genome functionality, including successful replication in two ecologically different hosts, is determined by the acquisition of new structural RNA elements.
3. Cyclization of the Flavivirus Genome
From a structural point of view, one of the most interesting and complex flavivirus phenomena is the acquisition of a circular closed-loop topology by the RNA genome (the cyclization process) [38]. The acquisition of this conformation was revealed in 1987 [29], but electron microscopy had already recorded the same behavior in alphaviruses during the 1970s [61]. Many unrelated viruses have since been shown to do the same [62][63][64][65][66][67]. In flaviviruses, genome cyclization is necessary for replication [6][11][23][68][69]; it ensures the synthesis of full-length genomes [29]. It also provides a control mechanism for translation [31][70] and infectivity [71] and helps to govern transitions between the different steps of the viral cycle [72].
In flaviviruses, cyclization depends on the establishment of non-covalent interactions between complementary sequences at the 5' and the 3' ends of the genome [73]. In MBFV, three pairs of complementary sequence motifs are responsible:
(i) The cyclization sequence 5'CS [29][39][74] located at the 5' end of the ORF, which pairs with the complementary 3'CS motif (also named CS1) at the base of the 3'SL (Figures 1 and 2). The complementarity between these sequence motifs has been demonstrated by different authors [11][23][74][75] who also showed that the preservation of base-pairing, independent of the actual nucleotides present, was enough to ensure efficient viral replication. The length of this sequence was found to be 10, 11 and 18 nt for the members of the DENV, JEV and YFV clades, respectively [11][76]. In the TBFV group, two pairs of cyclization sequences—5'-3'CS-A and 5'-3'CS-B—have been recorded (Figure 2). The CS-B pair is located in a position analogous to the CS motifs in MBFV, whereas the CS-A pair is displaced to the ends of the genome: thus, the 5' CS-A sequence lies upstream of the AUG codon and the 3'CS-A is located at the base of 3'SL (Figure 3) [39][74]. Both pairs differ in sequence from the CS motifs described in MBFV [77].
(ii) The 5' downstream sequence of the AUG region, 5'DAR, and its partner 3'DAR, which overlaps with the sHP element (and therefore the 3'CS motif) in the linear conformation of viral RNA (Figure 2) [69][78][79]. The formation of the DAR pair is essential for virus replication, but mismatches or low complementarity between the 5'DAR and the 3'DAR are better tolerated in mammalian than in mosquito cells [69]. Thus, the DAR interaction is subject to differential selection pressure and contributes to host adaptation [21][69]. Interestingly, with the exception of the clades YFV, SPOV and DENV, the remaining MBFV members contain two pairs of DAR sequences, DARI and DARII (Figure 2) [28]. In addition, while 3'DAR motifs seem to be conserved across flaviviruses, the 5'DAR sequences are only conserved across DENV serotypes [78].
(iii) The 5' upstream sequence of the AUG region, 5'UAR, and its complementary sequence 3'UAR within the basal portion of the stem in the 3'SL element (Figure 2) [11][12]. The formation of the so-called UAR pair requires the unwinding of the UFS at the 5'UTR, which is likely mediated by cellular factors such as AUF-1 [58]. Destabilization at the base of 3'SL within the 3'UTR is also needed (Figures 1 and 2) [12][13][59][80].
The compilation of functional data [20][80][81][82][83] has led to a cyclization model being proposed in which the initial interaction involving the CS and DAR motifs induces the unwinding of the sHP at the base of the 3'SL, thus releasing the 3'UAR sequence to form the UAR pair. The 5'UAR-3'UAR interaction would then stabilize the tertiary structure and promote high-order structural changes [72]. In TBFV, genome cyclization is further stabilized by an additional kissing-loop interaction between the conserved hexanucleotide motif of the 5'SL6 element and its complementary sequence at the 3'UTR (3'SL3) (Figures 1 and 2) [84].
The cyclization process can be facilitated by additional cis-structural elements. For example, different MBFVs form a three-stemmed PK downstream of the 5'CS (DCS-PK) (Figures 1 and 2) [85], which, together with the UAR, DAR and CS pairs, generates a functional domain that regulates the conformational switch inducing the cyclization of the genome. Interestingly, this region is highly conserved in MBFVs, with the exception of the YFV clade, in which a single hairpin has substituted the DCS-PK (Figure 1). Whether this hairpin performs an equivalent function to that shown by DCS-PK remains to be seen.
Genome cyclization may fit into the intracellular viral infection process as follows: in its linear form, the actively translated RNA genome presents the UFS duplex, which is crucial for NS5 recruitment to the SLA element. This induces a translationally repressed state [13]. Genome cyclization then occurs via the establishment of long-distance RNA–RNA interactions, and the recruitment of viral proteins and cellular factors [56][58][81][86][87][88]. The UFS then unwinds. This conformational effect promotes the transfer of the NS5 to the free 3' end, initiating the replication of the circular form [9][44][89]. The UFS may therefore be considered a switch for controlling the transition from viral translation to replication. It has also been proposed that the UFS acts as a structurally dynamic element to control NS5 recruitment during replication in a manner dependent on viral RNA levels [90], but this needs to be confirmed.
4. Subgenomic Flavivirus RNAs
In addition to the functional roles of the discrete 3'UTR elements in the formation of the secondary and tertiary structures described above, the 3'UTR participates in the production of small, flavivirus-specific, non-coding RNAs known as sfRNAs. sfRNAs result from the incomplete degradation of the viral RNA genome; they accumulate in the cytoplasm of infected cells [91].
4.1. Biogenesis of sfRNAs
Uncapped viral genomic RNAs are digested by the 5'-3' exoribonuclease XRN1 in mammalian cells, or by its homolog Pacman in insect cells [26][92]. This digestion ends, however, if the enzyme encounters specific 3D structures in the 3'UTR known as XRN1-resistant RNA structures (xrRNAs), thus producing sfRNAs. The amounts of different sfRNAs produced, and the function of these different types, varies between flaviviruses. Host alternation during the viral cycle also determines the production of different sfRNA species [93][94].
The members of the MBFV group show differences in their production of sfRNAs. Members of the JEV clade bear the SL-II element, which participates in the formation of xrRNA1; when XRN1 encounters the latter, digestion stops and sfRNA1 is produced [26]. Other sfRNAs are produced as a result of additional xrRNA elements downstream of SL-II, indicating that the presence of duplicated elements leads to the duplication of the tertiary ‘stop’ structures. The appearance of shorter sfRNAs also indicates that XRN1 is not completely blocked by the xrRNA1 site [15]. Indeed, three additional species of sfRNAs (sfRNA2, sfRNA3 and sfRNA4) have been described in WNV, the result of XRN1 stalling at xrRNA structures involving the SL-IV (xrRNA2), 5'DB (xrRNA3) and 3'DB (xrRNA4) structural elements, respectively [26][32][95]. In contrast, only two sfRNAs (sfRNA1 and 2), have been identified in human-adapted DENV, the result of XRN1 stalling at SL-II- and SL-IV-dependent xrRNA sites, respectively [94][96]. A similar pattern has been seen in ZIKV-infected mosquito and human cells [97], although a third sfRNA (200 nt-long) species might also be produced in mosquitoes [98]. Unlike that seen for the JEV and DENV clades, the members of YFV produce two sfRNAs via the influence of a single xrRNA involving the SL-E structural element. Both of these sfRNAs share the 5' end but vary in the 3' end, as sfRNA2 lacks the 3'SL; the mechanism behind its generation is unknown, as is its function [93][99]. So far, such truncated sfRNAs have only been observed in YFV [100]. Two sfRNAs have been reported in TBEV, and a single sfRNA has been detected for the ISFV and NKFV group members [101][102][103].
Alternative pathways leading to the accumulation of sfRNAs have been proposed. One suggests the existence of promoter sequences close to the XRN1 stalling site, as seen in the 3'UTR of JEV clade members. In this case, subgenomic RNA production would derive from RNA transcription. This assumption is further supported by the observation that, in these viruses, sfRNAs accumulate even more strongly in XRN1-knock down cells [104]. A similar strategy might be followed by distantly related plant viruses of the family Tombusviridae [105], suggesting a convergence in the evolution of subgenomic RNA production systems among different viruses.
4.2. 3D xrRNA Folding Is Preserved across Flaviviruses
The overall folding of xrRNAs is well-conserved across flaviviruses, although significant variability in the primary and secondary structural elements can be found. Different sequence motifs and secondary structures can therefore induce the same three-dimensional conformation.
Several experimental strategies have been used to elucidate the architecture of the flavivirus 3'UTR xrRNA structures, the secondary structure elements, and the tertiary interactions involved in their formation. Although all xrRNAs share a common threedimensional structure [103][106], differences in the secondary structure [93][94] have led to two major classes of xrRNA structures being defined: (i) class 1, which comprises xrRNAs present in the 3'UTR of MBFV and ISFV members; and (ii) class 2, which includes those xrRNAs found in TBFV and NKFV members [101]. Some authors have contested this division, arguing that the tertiary interactions of the class 2 xrRNAs structures can be used to predict those of class 1 [103]. However, the results of a recent study combining bioinformatics, biochemical and structural analysis suggest the further redefinition of class 1 into two subclasses: 1a and 1b [107]. Although full consensus is yet to be reached, most authors agree that the use of the two-class classification system aids for a more comprehensive understanding of xrRNAs and provides a tool for searching for new xrRNAs in different viral systems.
All xrRNA structures found across the genus Flavivirus share three SLs that fold into a three-way junction [32][95][96][100][106]. These SLs are connected by RNA–RNA interactions to form a PK. Depending on the class of xrRNA, these interactions range from canonical Watson–Crick contacts to electrostatic stabilizations. In all cases, the xrRNA folds into a ring-like structure in which the 5' end of the RNA passes through it, forming a kind of knot. The helicase activity of XRN1 is unable to unfold this ring-like structure, leading to the blockage of viral genome degradation and the formation of sfRNAs [93][97][100][106]. In vitro assays have shown that the xrRNA ring-like structure also blocks 5'-3' viral RNA degradation performed by unrelated exoribonucleases [101]. However, it does not impede the advance of enzymes that work in the 3'-5' direction, such as viral NS5 polymerase [100][108]. Thus, xrRNA structures mechanically block XRN1 via the formation of a specific three dimensional structure rather than establishing specific interactions with any enzyme, or by promoting the thermodynamic stabilization of the xrRNA [101].
A clear example of how different primary and secondary structures can lead to the same 3D xrRNA has been reported in the divergent flavivirus Tamana bat virus (TABV) [30][107][109][110]. X-ray crystallization studies have shown that while the ring-like structure of the xrRNA is preserved across TABV and other flaviviruses, significant variations occur in the corresponding nucleotide sequences and secondary structural elements [107][110]. One major difference resides in the shortening of the stems that form the ring-like structure. In order to accommodate the required tertiary interactions associated with these shorter helices, the overall structure is bent, a feature not seen in MBFV xrRNAs with longer stems. Consequently, in TABV, the xrRNA is more compact and rigid. In addition, the region encircling the 5' end of the xrRNA lacks Watson–Crick pairs, requiring the interplay of Mg2+ ions and water molecules to stabilize the final structure [110]. These differences support the idea that the TABV represents a phylogenetically distinct lineage within flaviviruses and mark the path for understanding the mechanisms that drive the generation of subgenomic RNAs in other virus families (see below). The characterization of the TABV’s xrRNAs provides additional structural information for searching new stalling sites in other RNA viruses.
4.3. The Role of sfRNAs
The requirement of XRN1 for the biogenesis of sfRNAs involves the shortage of this exoribonuclease for physiological cellular mRNA turnover, promoting cytopathic effects [26][111][112]. Cytopathicity also occurs through the direct inhibition of the Bcl-2 protein by sfRNAs.
sfRNAs are also involved in strategies that interfere with antiviral responses. One of these strategies involves the recognition and binding of isoforms of the protein Dicer along with protein components of the interference machinery. Thus, proteins essential to the cell for the physiological production of miRNAs are sequestered. This also prevents the generation of siRNAs from double-stranded regions of the flavivirus genome [113]. Flaviviruses can also evade the type I interferon-mediated response. This involves the use of different viral proteins, as described for other members of the family Flaviviridae. It also occurs via the direct sequestration by sfRNAs of cellular factors—such as TRIM25, G3BP1/2 and CAPRIN1—that participate in the interferon response pathway [114]. Protein recruitment may require specific sequence motifs within the 3'UTR, but given the variation shown by this region, other unknown mechanisms likely help impair the interferon-mediated response.
The importance of sfRNAs as driving elements in the evolution of flaviviruses has been reviewed by Slonchak et al. (2018). The accumulation of mutations in specific domains within the 3'UTR modifies the genomic:subgenomic RNA ratio; larger numbers of sfRNA may help to promote outbreaks of disease [100]. It should also be remembered that duplicated secondary structural elements within the 3'UTR account for specific sfRNA production patterns in different hosts, helping them to switch between different natural hosts [24][93].
References
- Blitvich, B.J.; Firth, A.E.; Kuhn, J.H. A review of flaviviruses that have no known arthropod vector. Viruses 2017, 9, 154.
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV virus taxonomy profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3.
- Brinton, M.A. The Molecular Biology of West Nile Virus: A new invader of the western hemisphere. Annu. Rev. Microbiol. 2002, 56, 371–402.
- Rice, C.M.; Lenches, E.M.; Eddy, S.R.; Shin, S.J.; Sheets, R.L.; Strauss, J.H. Nucleotide sequence of yellow fever virus: Implications or flavivirus gene expression and evolution. Science 1985, 229, 726–733.
- Fernández-Sanlés, A.; Ríos-Marco, P.; Romero-López, C.; Berzal-Herranz, A. Functional information stored in the conserved structural RNA domains of flavivirus genomes. Front. Microbiol. 2017, 8, 546.
- Ng, W.C.; Soto-Acosta, R.; Bradrick, S.S.; Garcia-Blanco, M.A.; Ooi, E.E. The 5ʹ and 3ʹ untranslated regions of the flaviviral genome. Viruses 2017, 9, 137.
- Markoff, L. 5′- and 3′-noncoding regions in flavivirus RNA. Adv. Virus Res. 2003, 59, 177–228.
- Dong, H.; Zhang, B.; Shi, P.Y. Terminal structures of West Nile virus genomic RNA and their interactions with viral NS5 protein. Virology 2008, 381, 123–135.
- Filomatori, C.V.; Lodeiro, M.F.; Alvarez, D.E.; Samsa, M.M.; Pietrasanta, L.; Gamarnik, A.V. A 5′ RNA element promotes Dengue virus RNA synthesis on a circular genome. Genes Dev. 2006, 20, 2238–2249.
- Lodeiro, M.F.; Filomatori, C.V.; Gamarnik, A.V. Structural and functional studies of the promoter element for Dengue virus RNA replication. J. Virol. 2009, 83, 993–1008.
- Alvarez, D.E.; Lodeiro, M.F.; Ludueña, S.J.; Pietrasanta, L.I.; Gamarnik, A.V. Long-range RNA-RNA interactions circularize the Dengue virus genome. J. Virol. 2005, 79, 6631–6643.
- Alvarez, D.E.; Filomatori, C.V.; Gamarnik, A.V. Functional analysis of Dengue virus cyclization sequences located at the 5′ and 3′UTRs. Virology 2008, 375, 223–235.
- Liu, Z.Y.; Li, X.F.; Jiang, T.; Deng, Y.Q.; Ye, Q.; Zhao, H.; Yu, J.Y.; Qin, C.F. Viral RNA switch mediates the dynamic control of flavivirus replicase recruitment by genome cyclization. Elife 2016, 5, e17636.
- Sharma, S.; Varani, G. NMR structure of Dengue and West Nile viruses stem-loop B: A key cis-acting element for flavivirus replication. Biochem. Biophys. Res. Commun. 2020, 531, 522–527.
- Göertz, G.P.; Abbo, S.R.; Fros, J.J.; Pijlman, G.P. Functional RNA during Zika virus infection. Virus Res. 2018, 254, 41–53.
- Mutebi, J.-P.; Rijnbrand, R.C.A.; Wang, H.; Ryman, K.D.; Wang, E.; Fulop, L.D.; Titball, R.; Barrett, A.D.T. Genetic relationships and evolution of genotypes of yellow fever virus and other members of the yellow fever virus group within the Flavivirus genus based on the 3′ noncoding region. J. Virol. 2004, 78, 9652–9665.
- Gritsun, T.S.; Gould, E.A. Origin and evolution of flavivirus 5′UTRs and panhandles: Trans-terminal duplications? Virology 2007, 366, 8–15.
- Khromykh, A.A.; Westaway, E.G.; Sakzewski, A. Completion of kunjin virus RNA sequence and recovery of an infectious RNA transcribed from stably cloned full-length cDNA. J. Virol. 1994, 68, 4580–4588.
- Proutski, V.; Gould, E.A.; Holmes, E.C. Secondary structure of the 3’ untranslated region of flaviviruses: Similarities and differences. Nucleic Acids Res. 1997, 25, 1194–1202.
- de Borba, L.; Villordo, S.M.; Marsico, F.L.; Carballeda, J.M.; Filomatori, C.V.; Gebhard, L.G.; Pallarés, H.M.; Lequime, S.; Lambrechts, L.; Vargas, I.S.; et al. RNA structure duplication in the Dengue virus 3´ UTR: Redundancy or host specificity? MBio 2019, 10.
- Villordo, S.M.; Filomatori, C.V.; Sánchez-Vargas, I.; Blair, C.D.; Gamarnik, A.V. Dengue virus RNA structure specialization facilitates host adaptation. PLoS Pathog. 2015, 11, 1–22.
- Villordo, S.M.; Gamarnik, A.V. Differential RNA sequence requirement for Dengue virus replication in mosquito and mammalian cells. J. Virol. 2013, 87, 9365–9372.
- Lo, M.K.; Tilgner, M.; Bernard, K.A.; Shi, P.-Y. Functional analysis of mosquito-borne flavivirus conserved sequence elements within 3′ untranslated region of West Nile Virus by use of a reporting replicon that differentiates between viral translation and RNA replication. J. Virol. 2003, 77, 10004–10014.
- Villordo, S.M.; Carballeda, J.M.; Filomatori, C.V.; Gamarnik, A.V. RNA structure duplications and flavivirus host adaptation. Trends Microbiol. 2016, 24, 270–283.
- Sztuba-Solinska, J.; Teramoto, T.; Rausch, J.W.; Shapiro, B.A.; Padmanabhan, R.; Le Grice, S.F.J. Structural complexity of Dengue virus untranslated regions: Cis-acting RNA motifs and pseudoknot interactions modulating functionality of the viral genome. Nucleic Acids Res. 2013, 41, 5075–5089.
- Pijlman, G.P.; Funk, A.; Kondratieva, N.; Leung, J.; Torres, S.; van der Aa, L.; Liu, W.J.; Palmenberg, A.C.; Shi, P.Y.; Hall, R.A.; et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008, 4, 579–591.
- Proutski, V.; Gaunt, M.W.; Gould, E.A.; Holmes, E.C. Secondary structure of the 3’-untranslated region of yellow fever virus: Implications for virulence, attenuation and vaccine development. J. Gen. Virol. 1997, 78, 1543–1549.
- Zeng, M.; Duan, Y.; Zhang, W.; Wang, M.; Jia, R.; Zhu, D.; Liu, M.; Zhao, X.; Yang, Q.; Wu, Y.; et al. Universal RNA secondary structure insight into mosquito-borne flavivirus (MBFV) cis-Acting RNA biology. Front. Microbiol. 2020, 11, 473.
- Hahn, C.S.; Hahn, Y.S.; Rice, C.M.; Lee, E.; Dalgarno, L.; Strauss, E.G.; Strauss, J.H. Conserved elements in the 3′ untranslated region of flavivirus RNAs and potential cyclization sequences. J. Mol. Biol. 1987, 198, 33–41.
- Ochsenreiter, R.; Hofacker, I.L.; Wolfinger, M.T. Functional RNA structures in the 3’UTR of tick-borne, insect-specific and no-known-vector flaviviruses. Viruses 2019, 11, 298.
- Manzano, M.; Reichert, E.D.; Polo, S.; Falgout, B.; Kasprzak, W.; Shapiro, B.A.; Padmanabhan, R. Identification of cis-acting elements in the 3′-untranslated region of the Dengue virus type 2 RNA that modulate translation and replication. J. Biol. Chem. 2011, 286, 22521–22534.
- Funk, A.; Truong, K.; Nagasaki, T.; Torres, S.; Floden, N.; Melian, E.B.; Edmonds, J.; Dong, H.; Shi, P.-Y.; Khromykh, A.A. RNA structures required for production of subgenomic flavivirus RNA. J. Virol. 2010, 84, 11407–11417.
- Olsthoorn, R.C.L.; Bol, J.F. Sequence comparison and secondary structure analysis of the 3′ noncoding region of flavivirus genomes reveals multiple pseudoknots. RNA 2001, 7, 1370–1377.
- Akiyama, B.M.; Graham, M.E.; O′donoghue, Z.; David Beckham, J.; Kieft, J.S. Three-dimensional structure of a flavivirus dumbbell RNA reveals molecular details of an RNA regulator of replication. bioRxiv 2020.
- Brinton, M.A.; Fernandez, A.V.; Dispoto, J.H. The 3′-nucleotides of flavivirus genomic RNA form a conserved secondary structure. Virology 1986, 153, 113–121.
- Gritsun, T.S.; Venugopal, K.; Zanotto, P.M.D.A.; Mikhailov, M.V.; Sall, A.A.; Holmes, E.C.; Polkinghorne, I.; Frolova, T.V.; Pogodina, V.V.; Lashkevich, V.A.; et al. Complete sequence of two tick-borne flaviviruses isolated from Siberia and the UK: Analysis and significance of the 5’ and 3’-UTRs. Virus Res. 1997, 49, 27–39.
- Gritsun, D.J.; Jones, I.M.; Gould, E.A.; Gritsun, T.S. Molecular archaeology of Flaviviridae untranslated regions: Duplicated RNA structures in the replication enhancer of flaviviruses and pestiviruses emerged via convergent evolution. PLoS ONE 2014, 9, e92056.
- Villordo, S.M.; Gamarnik, A.V. Genome cyclization as strategy for flavivirus RNA replication. Virus Res. 2009, 139, 230–239.
- Mandl, C.W.; Holzmann, H.; Kunz, C.; Heinz, F.X. Complete genomic sequence of Powassan virus: Evaluation of genetic elements in tick-borne versus mosquito-borne flaviviruses. Virology 1993, 194, 173–184.
- Charlier, N.; Leyssen, P.; Pleij, C.W.A.; Lemey, P.; Billoir, F.; Van Laethem, K.; Vandamme, A.-M.; De Clercq, E.; de Lamballerie, X.; Neyts, J. Complete genome sequence of Montana Myotis leukoencephalitis virus, phylogenetic analysis and comparative study of the 3′ untranslated region of flaviviruses with no known vector. J. Gen. Virol. 2002, 83, 1875–1885.
- Khromykh, A.A.; Kondratieva, N.; Sgro, J.-Y.; Palmenberg, A.; Westaway, E.G. Significance in replication of the terminal nucleotides of the flavivirus genome. J. Virol. 2003, 77, 10623–10629.
- Elghonemy, S.; Davis, W.G.; Brinton, M.A. The majority of the nucleotides in the top loop of the genomic 3′ terminal stem loop structure are cis-acting in a West Nile virus infectious clone. Virology 2005, 331, 238–246.
- Tilgner, M.; Deas, T.S.; Shi, P.Y. The flavivirus-conserved penta-nucleotide in the 3′ stem-loop of the West Nile virus genome requires a specific sequence and structure for RNA synthesis, but not for viral translation. Virology 2005, 331, 375–386.
- Filomatori, C.V.; Iglesias, N.G.; Villordo, S.M.; Alvarez, D.E.; Gamarnik, A.V. RNA sequences and structures required for the recruitment and activity of the Dengue virus polymerase. J. Biol. Chem. 2011, 286, 6929–6939.
- Holden, K.L.; Harris, E. Enhancement of Dengue virus translation: Role of the 3′ untranslated region and the terminal 3′ stem-loop domain. Virology 2004, 329, 119–133.
- Holden, K.L.; Stein, D.A.; Pierson, T.C.; Ahmed, A.A.; Clyde, K.; Iversen, P.L.; Harris, E. Inhibition of Dengue virus translation and RNA synthesis by a morpholino oligomer targeted to the top of the terminal 3′ stem-loop structure. Virology 2006, 344, 439–452.
- Hodge, K.; Tunghirun, C.; Kamkaew, M.; Limjindaporn, T.; Yenchitsomanus, P.T.; Chimnaronk, S. Identification of a conserved RNA-dependent RNA polymerase (RdRp)-RNA interface required for flaviviral replication. J. Biol. Chem. 2016, 291, 17437–17449.
- Blackwell, J.L.; Brinton, M.A. BHK cell proteins that bind to the 3’ stem-loop structure of the West Nile virus genome RNA. J. Virol. 1995, 69, 5650–5658.
- Blackwell, J.L.; Brinton, M.A. Translation elongation factor-1 alpha interacts with the 3’ stem-loop region of West Nile virus genomic RNA. J. Virol. 1997, 71, 6433–6444.
- Davis, W.G.; Blackwell, J.L.; Shi, P.-Y.; Brinton, M.A. Interaction between the cellular protein eEF1A and the 3′-terminal stem-loop of West Nile virus genomic RNA facilitates viral minus-strand RNA synthesis. J. Virol. 2007, 81, 10172–10187.
- Li, W.; Li, Y.; Kedersha, N.; Anderson, P.; Emara, M.; Swiderek, K.M.; Moreno, G.T.; Brinton, M.A. Cell proteins TIA-1 and TIAR interact with the 3′ stem-loop of the West Nile virus complementary minus-strand RNA and facilitate virus replication. J. Virol. 2002, 76, 11989–12000.
- Emara, M.M.; Brinton, M.A. Interaction of TIA-1/TIAR with West Nile and Dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 9041–9046.
- Albornoz, A.; Carletti, T.; Corazza, G.; Marcello, A. The stress granule component TIA-1 binds tick-borne encephalitis virus RNA and is recruited to perinuclear sites of viral replication to inhibit viral translation. J. Virol. 2014, 88, 6611–6622.
- Paranjape, S.M.; Harris, E. Y box-binding protein-1 binds to the Dengue virus 3′-untranslated region and mediates antiviral effects. J. Biol. Chem. 2007, 282, 30497–30508.
- Gomila, R.C.; Martin, G.W.; Gehrke, L. NF90 binds the Dengue virus RNA 3′ terminus and is a positive regulator of Dengue virus replication. PLoS ONE 2011, 6, e16687.
- García-Montalvo, B.M.; Medina, F.; Del Angel, R.M. La protein binds to NS5 and NS3 and to the 5′ and 3′ ends of Dengue 4 virus RNA. Virus Res. 2004, 102, 141–150.
- Vashist, S.; Anantpadma, M.; Sharma, H.; Vrati, S. La protein binds the predicted loop structures in the 3′ non-coding region of Japanese encephalitis virus genome: Role in virus replication. J. Gen. Virol. 2009, 90, 1343–1352.
- Friedrich, S.; Engelmann, S.; Schmidt, T.; Szczepankiewicz, G.; Bergs, S.; Liebert, U.G.; Kümmerer, B.M.; Golbik, R.P.; Behrens, S.-E. The host factor AUF1 p45 supports flavivirus propagation by triggering the RNA switch required for viral genome cyclization. J. Virol. 2018, 92, e01647-17.
- Meyer, A.; Freier, M.; Schmidt, T.; Rostowski, K.; Zwoch, J.; Lilie, H.; Behrens, S.E.; Friedrich, S. An RNA thermometer activity of the West Nile virus genomic 3’-terminal stem-loop element modulates viral replication efficiency during host switching. Viruses 2020, 12, 104.
- Davis, W.G.; Basu, M.; Elrod, E.J.; Germann, M.W.; Brinton, M.A. Identification of cis-acting nucleotides and a structural feature in West Nile virus 3’-terminus RNA that facilitate viral minus strand RNA synthesis. J. Virol. 2013, 87, 7622–7636.
- Hsu, M.T.; Kung, H.J.; Davidson, N. An electron microscope study of Sindbis virus RNA. Cold Spring Harb. Symp. Quant. Biol. 1974, 38, 943–950.
- Flick, R.; Hobom, G. Transient bicistronic vRNA segments for indirect selection of recombinant influenza viruses. Virology 1999, 262, 93–103.
- Kohl, A.; Dunn, E.F.; Lowen, A.C.; Elliott, R.M. Complementarity, sequence and structural elements within the 3′ and 5′ non-coding regions of the Bunyamwera orthobunyavirus S segment determine promoter strength. J. Gen. Virol. 2004, 85, 3269–3278.
- Mir, M.A.; Panganiban, A.T. The hantavirus nucleocapsid protein recognizes specific features of the viral RNA panhandle and is altered in conformation upon RNA binding. J. Virol. 2005, 79, 1824–1835.
- Mir, M.A.; Brown, B.; Hjelle, B.; Duran, W.A.; Panganiban, A.T. Hantavirus N protein exhibits genus-specific recognition of the viral RNA panhandle. J. Virol. 2006, 80, 11283–11292.
- Perez, M.; de la Torre, J.C. Characterization of the genomic promoter of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2003, 77, 1184–1194.
- Patton, J.T. Rotavirus RNA replication and gene expression. Novartis Found. Symp. 2001, 238, 64–81.
- Men, R.; Bray, M.; Clark, D.; Chanock, R.M.; Lai, C.J. Dengue type 4 virus mutants containing deletions in the 3’ noncoding region of the RNA genome: Analysis of growth restriction in cell culture and altered viremia pattern and immunogenicity in rhesus monkeys. J. Virol. 1996, 70, 3930–3937.
- Li, X.-D.; Deng, C.-L.; Yuan, Z.-M.; Ye, H.-Q.; Zhang, B. Different degrees of 5’-to-3’ DAR interactions modulate Zika virus genome cyclization and host-specific replication. J. Virol. 2019, 94.
- Wei, Y.; Qin, C.; Jiang, T.; Li, X.; Zhao, H.; Liu, Z.; Deng, Y.; Liu, R.; Chen, S.; Yu, M.; et al. Translational regulation by the 3′ untranslated region of the Dengue type 2 virus genome. Am. J. Trop. Med. Hyg. 2009, 81, 817–824.
- Proutski, V.; Gritsun, T.S.; Gould, E.A.; Holmes, E.C. Biological consequences of deletions within the 3’-untranslated region of flaviviruses may be due to rearrangements of RNA secondary structure. Virus Res. 1999, 64, 107–123.
- Sanford, T.J.; Mears, H.V.; Fajardo, T.; Locker, N.; Sweeney, T.R. Circularization of flavivirus genomic RNA inhibits de novo translation initiation. Nucleic Acids Res. 2019, 47, 9789–9802.
- Garcia-Blanco, M.A.; Vasudevan, S.G.; Bradrick, S.S.; Nicchitta, C. Flavivirus RNA transactions from viral entry to genome replication. Antivir. Res. 2016, 134, 244–249.
- Khromykh, A.A.; Meka, H.; Guyatt, K.J.; Westaway, E.G. Essential role of cyclization sequences in flavivirus RNA replication. J. Virol. 2001, 75, 6719–6728.
- Zhang, B.; Dong, H.; Zhou, Y.; Shi, P.-Y. Genetic interactions among the West Nile virus methyltransferase, the RNA-dependent RNA polymerase, and the 5′ stem-loop of genomic RNA. J. Virol. 2008, 82, 7047–7058.
- Corver, J.; Lenches, E.; Smith, K.; Robison, R.A.; Sando, T.; Strauss, E.G.; Strauss, J.H. Fine mapping of a cis-acting sequence element in Yellow fever virus RNA that is required for RNA replication and cyclization. J. Virol. 2003, 77, 2265–2270.
- Kofler, R.M.; Hoenninger, V.M.; Thurner, C.; Mandl, C.W. Functional analysis of the tick-borne encephalitis virus cyclization elements indicates major differences between mosquito-borne and tick-borne flaviviruses. J. Virol. 2006, 80, 4099–4113.
- Friebe, P.; Harris, E. Interplay of RNA elements in the Dengue virus 5′ and 3′ ends required for viral RNA replication. J. Virol. 2010, 84, 6103–6118.
- Friebe, P.; Shi, P.-Y.; Harris, E. The 5’ and 3’ downstream AUG region elements are required for mosquito-borne flavivirus RNA replication. J. Virol. 2011, 85, 1900–1905.
- Zhang, B.; Dong, H.; Stein, D.A.; Iversen, P.L.; Shi, P.Y. West Nile virus genome cyclization and RNA replication require two pairs of long-distance RNA interactions. Virology 2008, 373, 1–13.
- Friedrich, S.; Schmidt, T.; Schierhorn, A.; Lilie, H.; Szczepankiewicz, G.; Bergs, S.; Liebert, U.G.; Golbik, R.P.; Behrens, S.E. Arginine methylation enhances the RNA chaperone activity of the West Nile virus host factor AUF1 p45. RNA 2016, 22, 1574–1591.
- Friedrich, S.; Schmidt, T.; Geissler, R.; Lilie, H.; Chabierski, S.; Ulbert, S.; Liebert, U.G.; Golbik, R.P.; Behrens, S.-E. AUF1 p45 promotes West Nile virus replication by an RNA chaperone activity that supports cyclization of the viral genome. J. Virol. 2014, 88, 11586–11599.
- de Borba, L.; Villordo, S.M.; Iglesias, N.G.; Filomatori, C.V.; Gebhard, L.G.; Gamarnik, A.V. Overlapping local and long-range RNA-RNA interactions modulate Dengue virus genome cyclization and replication. J. Virol. 2015, 89, 3430–3437.
- Tsetsarkin, K.A.; Liu, G.; Shen, K.; Pletnev, A.G. Kissing-loop interaction between 5′ and 3′ ends of tick-borne Langat virus genome “bridges the gap” between mosquito-and tick-borne flaviviruses in mechanisms of viral RNA cyclization: Applications for virus attenuation and vaccine development. Nucleic Acids Res. 2016, 44, 3330–3350.
- Liu, Z.-Y.; Zhao, H. Novel cis-acting element within the capsid-coding region enhances flavivirus viral-RNA replication by regulating genome cyclization. J. Virol. 2013, 87, 6804–6818.
- Ivanyi-Nagy, R.; Darlix, J.L. Core protein-mediated 5’-3’ annealing of the West Nile virus genomic RNA in vitro. Virus Res. 2012, 169, 448–457.
- Gebhard, L.G.; Kaufman, S.B.; Gamarnik, A.V. Novel ATP-independent RNA annealing activity of the Dengue virus NS3 helicase. PLoS ONE 2012, 7, e36244.
- Vashist, S.; Bhullar, D.; Vrati, S. La protein can simultaneously bind to both 3′- and 5′-noncoding regions of Japanese encephalitis virus genome. DNA Cell Biol. 2011, 30, 339–346.
- Fajardo, T.; Sanford, T.J.; Mears, H.V.; Jasper, A.; Storrie, S.; Mansur, D.S.; Sweeney, T.R. The flavivirus polymerase NS5 regulates translation of viral genomic RNA. Nucleic Acids Res. 2020, 48, 5081–5093.
- Liu, Z.Y.; Qin, C.F. Structure and function of cis-acting RNA elements of flavivirus. Rev. Med. Virol. 2020, 30, e2092.
- Roby, J.A.; Pijlman, G.P.; Wilusz, J.; Khromykh, A.A. Noncoding subgenomic flavivirus RNA: Multiple functions in West Nile virus pathogenesis and modulation of host responses. Viruses 2014, 6, 404–427.
- Jones, C.I.; Zabolotskaya, M.V.; Newbury, S.F. The 5′ → 3′ exoribonuclease XRN1/Pacman and its functions in cellular processes and development. Wiley Interdiscip. Rev. RNA 2012, 3, 455–468.
- Kieft, J.S.; Rabe, J.L.; Chapman, E.G. New hypotheses derived from the structure of a flaviviral XRN1-resistant RNA: Conservation, folding, and host adaptation. RNA Biol. 2015, 12, 1169–1177.
- Filomatori, C.V.; Carballeda, J.M.; Villordo, S.M.; Aguirre, S.; Pallaré, H.M.; Maestre, A.M.; Sánchez-Vargas, I.; Blair, C.D.; Fabri, C.; Morales, M.A.; et al. Dengue virus genomic variation associated with mosquito adaptation defines the pattern of viral non-coding RNAs and fitness in human cells. PLoS Pathog. 2017, 13, e1006265.
- Clarke, B.D.; Roby, J.A.; Slonchak, A.; Khromykh, A.A. Functional non-coding RNAs derived from the flavivirus 3’ untranslated region. Virus Res. 2015, 206, 53–61.
- Chapman, E.G.; Moon, S.L.; Wilusz, J.; Kieft, J.S. RNA structures that resist degradation by XRN1 produce a pathogenic Dengue virus RNA. Elife 2014, 3, e01892.
- Akiyama, B.M.; Laurence, H.M.; Massey, A.R.; Costantino, D.A.; Xie, X.; Yang, Y.; Shi, P.Y.; Nix, J.C.; Beckham, J.D.; Kieft, J.S. Zika virus produces noncoding RNAs using a multi-pseudoknot structure that confounds a cellular exonuclease. Science 2016, 354, 1148–1152.
- Slonchak, A.; Hugo, L.E.; Freney, M.E.; Hall-Mendelin, S.; Amarilla, A.A.; Torres, F.J.; Setoh, Y.X.; Peng, N.Y.G.; Sng, J.D.J.; Hall, R.A.; et al. Zika virus noncoding RNA suppresses apoptosis and is required for virus transmission by mosquitoes. Nat. Commun. 2020, 11, 1–14.
- Silva, P.A.G.C.; Pereira, C.F.; Dalebout, T.J.; Spaan, W.J.M.; Bredenbeek, P.J. An RNA pseudoknot is required for production of yellow fever virus subgenomic RNA by the host nuclease XRN1. J. Virol. 2010, 84, 11395–11406.
- Slonchak, A.; Khromykh, A.A. Subgenomic flaviviral RNAs: What do we know after the first decade of research. Antivir. Res. 2018, 159, 13–25.
- MacFadden, A.; Òdonoghue, Z.; Silva, P.A.G.C.; Chapman, E.G.; Olsthoorn, R.C.; Sterken, M.G.; Pijlman, G.P.; Bredenbeek, P.J.; Kieft, J.S. Mechanism and structural diversity of exoribonuclease-resistant RNA structures in flaviviral RNAs. Nat. Commun. 2018, 9, 1–11.
- Wastika, C.E.; Harima, H.; Sasaki, M.; Hang’ombe, B.M.; Eshita, Y.; Qiu, Y.; Hall, W.W.; Wolfinger, M.T.; Sawa, H.; Orba, Y. Discoveries of exoribonuclease-resistant structures of insect-specific flaviviruses isolated in Zambia. Viruses 2020, 12, 1017.
- Dilweg, I.W.; Bouabda, A.; Dalebout, T.; Gultyaev, A.P.; Bredenbeek, P.J.; Olsthoorn, R.C.L. XRN1-resistant RNA structures are well-conserved within the genus flavivirus. RNA Biol. 2020, 1–9.
- Chen, Y.-S.; Fan, Y.-H.; Tien, C.-F.; Yueh, A.; Chang, R.-Y. The conserved stem-loop II structure at the 3’ untranslated region of Japanese encephalitis virus genome is required for the formation of subgenomic flaviviral RNA. PLoS ONE 2018, 13, e0201250.
- Gunawardene, C.D.; Newburn, L.R.; Andrew White, K. A 212-nt long RNA structure in the Tobacco necrosis virus-D RNA genome is resistant to XRN degradation. Nucleic Acids Res. 2019, 47, 9329–9342.
- Chapman, E.G.; Costantino, D.A.; Rabe, J.L.; Moon, S.L.; Wilusz, J.; Nix, J.C.; Kieft, J.S. The structural basis of pathogenic subgenomic flavivirus RNA (sfRNA) production. Science 2014, 344, 307–310.
- Szucs, M.; Nichols, P.; Jones, R.; Vicens, Q.; Kieft, J. A new subclass of exoribonuclease resistant RNA found in multiple Flaviviridae genera. MBio 2020, 11, e02352-20.
- Charley, P.A.; Wilusz, J. Standing your ground to exoribonucleases: Function of Flavivirus long non-coding RNAs. Virus Res. 2016, 212, 70–77.
- de Lamballerie, X.; Crochu, S.; Billoir, F.; Neyts, J.; de Micco, P.; Holmes, E.C.; Gould, E.A. Genome sequence analysis of Tamana bat virus and its relationship with the genus Flavivirus. J. Gen. Virol. 2002, 83, 2443–2454.
- Jones, R.A.; Steckelberg, A.-L.; Vicens, Q.; Szucs, M.J.; Akiyama, B.M.; Kieft, J.S. Different tertiary interactions create the same important 3-D features in a distinct flavivirus xrRNA. RNA 2021, 27, 54–65.
- Moon, S.L.; Anderson, J.R.; Kumagai, Y.; Wilusz, C.J.; Akira, S.; Khromykh, A.A.; Wilusz, J. A noncoding RNA produced by arthropod-borne flaviviruses inhibits the cellular exoribonuclease XRN1 and alters host mRNA stability. RNA 2012, 18, 2029–2040.
- Liu, Y.; Liu, H.; Zou, J.; Zhang, B.; Yuan, Z. Dengue virus subgenomic RNA induces apoptosis through the Bcl-2-mediated PI3k/Akt signaling pathway. Virology 2014, 448, 15–25.
- Moon, S.L.; Dodd, B.J.T.; Brackney, D.E.; Wilusz, C.J.; Ebel, G.D.; Wilusz, J. Flavivirus sfRNA suppresses antiviral RNA interference in cultured cells and mosquitoes and directly interacts with the RNAi machinery. Virology 2015, 485, 322–329.
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.Z.; Tan, H.C.; Sessions, O.M.; Ward, A.M.; Gubler, D.J.; et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350, 217–221.

