 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Eugenia Gallardo | + 2690 word(s) | 2690 | 2021-04-14 10:12:05 | | | |
| 2 | Rita Xu | Meta information modification | 2690 | 2021-04-19 08:30:13 | | |
Video Upload Options
Neurodegenerative diseases (ND), including Alzheimer’s (AD) and Parkinson’s Disease (PD), are becoming increasingly more common and are recognized as a social problem in modern societies. These disorders are characterized by a progressive neurodegeneration and are considered one of the main causes of disability and mortality worldwide. Currently, there is no existing cure for AD nor PD and the clinically used drugs aim only at symptomatic relief, and are not capable of stopping neurodegeneration.
1. Introduction
The advances in medicine and the better quality of life of the general population have increased the average lifespan worldwide. According to a 2019 survey of the United Nations, 9% of the world’s population, the equivalent to 700 million people, is at/or above 65 years old and this number is expected to grow to at least 2 billion by 2050 [1]. Consequently, age-related diseases, where neurodegenerative conditions (ND) are included, are becoming more common and being recognized as a social problem in modern societies [2]. ND are characterized by a heterogeneous and progressive degeneration of the central and/or the peripheric nervous systems, as a consequence of the death of neuronal cells [3]. So far, hundreds of ND have been identified; however, each of them displays differences in terms of pathological characteristics, symptoms, and treatments [3]. Specifically, Alzheimer’s (AD) and Parkinson’s Disease (PD) are considered the most prevalent ND, affecting 11 and 5 in 1000 individuals more than 65 years old, respectively [4]. In fact, in 2020, more than 44 million and 10 million patients were diagnosed with AD and PD, respectively [4]. Despite the intense research and investment behind these disorders, their aetiology is still unknown, but it is thought to be caused by a combination of factors, such as the individual’s lifestyle and genetics, but also environmental factors, like exposure to toxins and pollution [4].
Indeed, several medical conditions, for instance primary neurological and neuropsychiatric diseases, are thought to contribute to dementia, mostly in elderly people, the age group with the highest dementia incidence. This condition can be developed as a consequence of some degree of degeneration, namely in the progression of the AD/PD diseases, vascular dementia, Lewy body accumulation, and frontotemporal lobar degeneration, among others. Moreover, mild cognitive impairment and dementia occurring across the individual’s lifespan might be related to chemotherapy-related cognitive dysfunction, vitamin deficiencies (e.g., B1, B12), normal pressure hydrocephalus, intracranial masses (e.g., subdural hematomas, brain tumors), traumatic brain injury, and psychiatric illness (depression major and anxiety) [5].
Unfortunately, there is no existing cure for these ND and the currently clinically used drugs aim only at symptomatic relief and are not capable of stopping neurodegeneration [6][7]. The biggest challenges in AD/PD drug development are the number of biological pathways and proteins involved in the diseases’ pathogenesis, the complexity of the affected organs (mostly the brain), and their aggressiveness [6][7]. Thus, the necessity of developing novel drugs has forced the pharmaceutical industry to employ new methodologies for the design of new compounds. The progresses in the biomolecular and structural fields allowed the determination of numerous three-dimensional (3D) structures of proteins, through nuclear magnetic resonance, X-ray crystallography, cryo-electron microscopy, among other techniques, and these have provided essential information about atomic interactions between protein-ligand [8]. The combination of computational and mathematical algorithms with the protein structural data has become increasingly used in modern medicinal chemistry to support drug design, generally defined as computer-aided drug design (CADD) [9]. These approaches can be subdivided into three main categories: sequence-based drug design, structure-based drug design (SBDD), and ligand-based drug design (LBDD) [9]. In particular, sequence-based drug design uses the protein sequence information deposited on the Protein Data Bank (PDB, https://www.rcsb.org/, accessed on 10 April 2021) database to build 3D homologue models of the protein 3D structure that can be further refined with molecular dynamics simulations [9]. The SBDD strategy is probably the most used in in silico studies; it uses the structural information found in the protein 3D structure to predict macromolecular binding sites and the affinity of ligands towards certain targets [9]. Within this strategy, molecular docking studies are the most known and used, including structure-based virtual screening, also known as target-based virtual screening, which allows for the selection of promising compounds from extensive libraries. Frequently, molecular dynamics are also used to obtain a more profound understanding of the interactions between ligands and macromolecules, as well as predicting the stability of the obtained binding poses. In most cases, a combination of these techniques is used in SBDD protocols [9]. Another methodology that is also commonly employed to study the affinity of ligands in targets that do not have a 3D structure available in the PDB is LBDD [9]. Similar to SBDD, this approach also allows the collection of the most relevant compounds from big libraries, and includes QSAR modeling, pharmacophore studies, similarity searching, among others [9]. Additionally, this approach can be combined with homology modeling [9]. These techniques have been improved over the years and are being increasingly used in drug development by researchers from academic centers and R&D of pharmaceutical companies, mostly in preliminary studies to select compounds with higher potential of success in further studies. Moreover, it is also possible to use software to filter molecules with positive ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties in addition to a predicted high affinity to the protein target [9].
2. Alzheimer’s Disease
AD is characterized by a slow and progressive decline in the cognitive functions and dementia, as a consequence of the loss of neurons, deterioration of the neurotransmission systems, and the accumulation of several proteins in the central nervous system (CNS) [10]. Overall, its pathophysiology features are the formation of amyloid plaques and neurofibrillary tangles in the brain, but more recently, dystrophic neurites, astrogliosis, neuropil threads, and microglial activation are also being reported [11]. Currently, only a few drugs are approved for clinical use in AD treatment and none of them can stop the progression of the disease, being mostly used in symptomatic therapy [12]. Despite all the scientific efforts made, more than 200 drug candidates have failed or been suspended from clinical trials in the last decade and no drug has been approved for AD treatment since 2003 [12]. These failures might be related to an inaccurate selection of the protein targets and the insufficient understanding of the complex etiology of AD [12]. In this topic, a characterization of the progresses accomplished in drug development employing computational approaches for the various protein targets involved in AD will be performed.
2.1. Acetylcholinesterase
Acetylcholinesterase (AChE, E.C. 3.1.1.7) is an enzyme involved in the termination of impulse transmission by rapid hydrolysis of acetylcholine into choline and acetic acid [13]. In AD, the patient’s cholinergic systems endure extensive degeneration changes, leading to a hypofunction of the cholinergic neurons and a decline in the endogenous levels of acetylcholine [14]. Hence, AChE inhibitors are administered to counteract these effects in an attempt to decrease the breakdown rate of acetylcholine and restore its synaptic levels [15]. To date, only three AChE inhibitors are used in AD therapy, donepezil, rivastigmine, and galantamine (Figure 1); however, they only offer symptomatic relief and are mostly used to treat mild to moderate dementia [7]. Thus, new and more effective AChE inhibitors are needed [16].
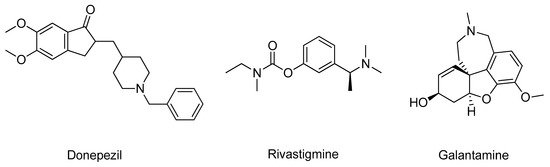
Figure 1. Structure of the clinically approved AChE inhibitors.
Many researchers took advantage of the available 3D human AChE structures deposited in the PDB database and the information regarding the interactions with several ligands to carry out in silico methodologies to design potential inhibitors [17]. Structurally, the target has two distinct binding sites: one is peripheral and is situated at the entrance of the gorge and the other is located in the catalytic site [15]. Currently, researchers are focused in molecules that can occupy both binding sites and inhibit acetylcholine hydrolysis [17]. Grafov et al. studied the affinity of several naturally occurring alkaloids for both binding sites of AChE (PDB#6H12) using neostigmine, a known AChE inhibitor, as positive control [18]. The molecular docking results demonstrated that the alkaloid 5-N-methylmaytenine (Figure 2) could simultaneously bind to both binding sites, mainly by hydrophobic interactions, and therefore, could have a high pharmacological potential towards the design of novel AChE inhibitors [18]. A similar analysis was carried out by Ortiz and his research group for other alkaloid compounds extracted from plants of the Hieronymiella genus, but employing an additional step of molecular dynamic simulations to evaluate the binding modes to better understand in vitro results [19]. In this study, the compound sanguinine (Figure 2), structurally similar to galantamine (Figure 1), showed the most promising binding energy and interacted with residues Trp84, Gly117, Glu199, Ser200, Phe330, and His440. In addition, it presented high in vitro inhibitory potency for AChE (PDB#1DX6) as well, which can indicate that these amino acids are essential to promote higher inhibition rates [19]. Mughal and co-workers synthetized a series of 4-thioflavonols that displayed very promising in vitro outputs and studied their interactions with the AChE (PDB#4BDT) active site through molecular docking [20]. Particularly, compound 1 (Figure 2) had the highest affinity and also formed identical interactions with the amino acids of the catalytic site of AChE similar to the observed for donepezil, especially with residues Trp86 and Tyr337 [20]. An identical strategy was performed to study other natural products, namely canadine derivatives [21], cinnamic acid derivatives, indolinones and cycloartane triterpenoids [22], phenolic acid derivatives [23], and synthetic compounds, such as arylisoxazole-phenylpiperazine derivatives [24], dipropargyl substituted diphenylpyrimidines [25], quinoline chalcone derivatives [26], and N-(4-methylpyridin-2-yl)thiophene-2-carboxamide analogs [27], as displayed in (Figure 2). Ranjan’s research group [28] studied the affinity of several organophosphate derivatives against AChE (PDB#1B41) using docking-based virtual screening combined with molecular dynamics simulations. The compounds were selected based on the interaction with the main residues of the catalytic triad, Ser203, Glu334, and His447 [28]. The top ranked ligand was phoxim ethyl phosphonate (Figure 2), displaying the highest binding energy with these residues and it was advanced for further in vitro studies [28].
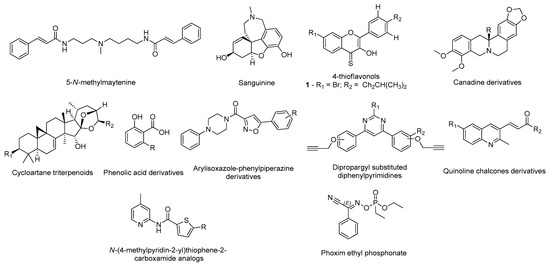
Figure 2. Structures of the studied AChE inhibitors.
Castro-Silva et al. studied the affinity of fucosterol (Figure 3) towards both binding sites of AChE (PDB#4EY7) and compared with the inhibitor neostigmine by docking and molecular dynamics to improve the analysis [29]. The results demonstrate that fucosterol has affinity towards both binding sites, by specifically interacting with the residues Trp286, Leu289, and Tyr341 of the peripheric site, and with the Trp86, Glu202, and Tyr449 of the AChE catalytic site, indicating that this compound might be a promising compound to advance for further studies [29]. Other researchers, namely Gurjar and co-workers, performed an in silico analysis in 2-substituted-4,5-diphenyl-1H-imidazole analogues (Figure 3) effecting a prediction of the compounds’ ADMET properties in addition to the molecular docking of the best ranked compounds, avoiding further testing of compounds with potential toxicity and unfavorable pharmacokinetics [30]. For instance, compound 2 (Figure 3) demonstrated the best results, being a potential candidate for further structural optimization for even better AChE inhibition [30]. Rocha and her group built and validated a machine learning model using pharmacophores based on the structures of more than 500 compounds with known and no inhibitory activity against AChE to predict the potential inhibitory activity of multiple indole alkaloids [31]. Of these, uleine (Figure 3) was predicted as being the alkaloid with the highest probability to present AChE inhibitory activity based on the in silico results [31]. The most promising compounds were further tested in vitro confirming the computational predictions regarding the AChE inhibition [31]. A distinct methodology was applied by Niu et al., building 2D- and 3D-QSAR models to classify molecules based on their potential to inhibit AChE, from a library of compounds that included known AChE inhibitors and non-inhibitors, with a predicted accuracy of 89.63% [32]. The most promising compounds were further tested by molecular docking to evaluate their affinity towards the target active site (PDB#1QTI) and the interaction with the residue Ser124 was demonstrated to be crucial for a higher affinity [32].
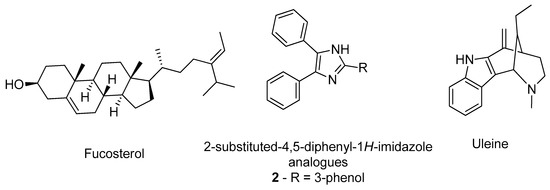
Figure 3. Structure of the studied AChE inhibitors.
2.2. N-Methyl-D-aspartate Receptor
The N-methyl-d-aspartate receptors (NMDAR) are a family of ligand-gated ionic membrane channels involved in non-selective cation transport and in the excitatory glutamatergic neurotransmission [33]. There are two types of NMDARs: the synaptic and the extra-synaptic receptors [33]. The synaptic are essential for synaptic plasticity and for the survival of neurons, while the extra-synaptic promote cell death and excitotoxicity, contributing for the etiology of AD [33]. The hyper-activation of the extra-synaptic NMDAR by glutamate, which is linked to an overproduction of free radicals and several enzymes that contribute to the deterioration of the CNS, can be controlled with NMDAR antagonists [34]. Currently, memantine (Figure 4) is the only NMDAR antagonist approved by the regulatory agencies for clinical use to treat moderate to severe dementia, selectively inhibiting the activity of extra-synaptic NMDARs [34]. In addition to its cognitive and functional pharmacological benefits, memantine has also demonstrated to slow down the emergence of further behavioral and psychotic manifestations. Furthermore, its administration resulted in significant increases in the extracellular concentrations of the neurotransmitters dopamine, norepinephrine, and their metabolites, demonstrating a biogenic amine neurotransmission enhancing effect useful in AD treatment [35].
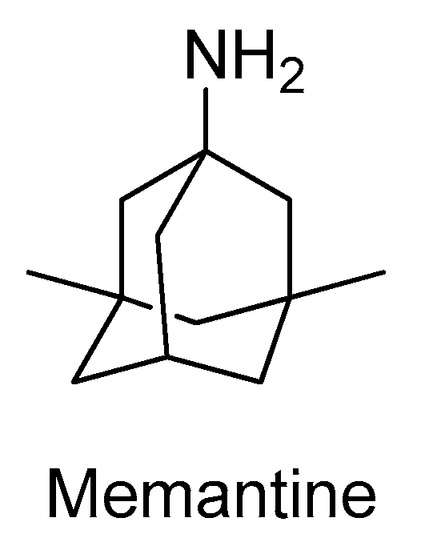
Figure 4. Structure of the clinically approved NMDAR antagonist.
Nevertheless, over the years, numerous studies reported the design of novel active NMDAR antagonists using in silico methodologies [9]. For instance, Ivanova and co-workers used a virtual screening approach to discover new potential NMDAR antagonists [36]. Using a combination of various machine learning methods, including artificial neural networks and advanced multilinear techniques to build QSAR models, they screened over 13,000 natural compounds and ranked them based on their predicted affinity towards the target (PDB#5TP9) [36]. The best candidates were also analysed by docking and molecular dynamics simulations to identify essential structural moieties that could serve as basis for the design and development of novel and improved NMDAR antagonists [36]. A distinct strategy was carried out by Sharma et al. using pharmacophore modeling and a four-phased virtual screening study to identify potential drug candidates [37]. The pharmacophore model was generated and validated with a library of 40 known NMDAR antagonists, followed by screening, where the compounds were sorted based on the Lipinski’s rules and in terms of affinity towards the target [37]. Additionally, the hits were submitted to a docking analysis to fully validate the used methodology [37]. The main residues in the NMDAR active site are His88, Ser114, Thr116, Art121, Gly172, Ser173, Thr174, and Tyr214, of which the predicted compound HTS 00987 (Figure 5) interacts with His88, Thr174, and Tyr214, while memantine was not predicted to interact with none of these residues, indicating a potential pharmacological interest of this compound [37]. Waqar et al. built a homologue model of the 3D structure of NMADR based on the structure of the rat NMDAR (PDB#3JPW) and analyzed by molecular docking the affinity of several conantokins towards this target [38]. Moreover, most of the compounds interacted with residues Gln110 and Glu236, in the NR2B subunit of the NMDAR, and with Ile111, Phe114, and Pro177 by hydrophobic interactions [38]. A similar binding pattern was observed for the rat crystal structure, indicating that conankotins might be potential NMDAR antagonists [38]. In another study carried out by Hu and co-workers, the affinity of several tetramethylpyrazine derivatives, with known NMDAR antagonist activity, was studied towards its catalytic site (PDB#5UOW) by molecular docking [39]. Specifically, compound 3 (Figure 5) demonstrated the most promising binding energy, as well as favorable interaction by hydrogen bonds with the amino acid Asn602 [39]. Kumar et al., studied the affinity of antipsychotic drugs towards NMDAR (PDB#1PBQ) by molecular docking to evaluate its potential use in the treatment of AD [40]. In the hydrophobic pocket of NMDAR 3D structure, the main residues are the Phe16, Phe92, Trp223, and Phe250 [40]. For the antipsychotic drug anisopirol (Figure 5), the compound of this group with the highest affinity mainly interacted with Phe92, Pro24, Thr126, Ser180, Trp223, and Phe246 [40]. Considering that this drug shared some interactions with the known NMDAR antagonist 5,7-dichlorokynurenic acid (DCKA), the co-crystallized ligand of this crystal structure, it might indicate that could also biologically interact with this target [40]. On the other hand, Singh et al. developed pharmacophore models based on the structure of ifenprodil, also a known NMDAR antagonist, performed virtual screening, studied the affinity of the hits by molecular docking and molecular dynamics simulations using the 3D structure of the NMDAR (PDB#5EWJ), as well as analyzed the hits ADMET properties [41]. The proposed study revealed that the molecules ZINC25726161 and ZINC95977857 (Figure 5) displayed a better affinity towards the target than the NMDAR antagonist drug ifenprodil, indicating that these virtual hits could have pharmacological interest [41].
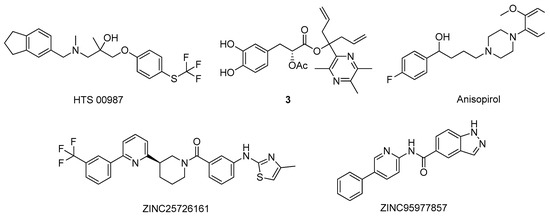
Figure 5. Structures of the studied NMDAR antagonists.
References
- World Population Ageing [highlights], (n.d.). Available online: (accessed on 2 March 2021).
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581.
- Van Bulck, M.; Sierra-Magro, A.; Alarcon-Gil, J.; Perez-Castillo, A.; Morales-Garcia, J. Novel Approaches for the Treatment of Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 719.
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480.
- Gale, S.A.; Acar, D.; Daffner, K.R. Dementia. Am. J. Med. 2018, 131, 1161–1169.
- McFarthing, K.; Buff, S.; Rafaloff, G.; Dominey, T.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2020. J. Parkinsons. Dis. 2020, 10, 757–774.
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6.
- Goodsell, D.S.; Zardecki, C.; Di Costanzo, L.; Duarte, J.M.; Hudson, B.P.; Persikova, I.; Segura, J.; Shao, C.; Voigt, M.; Westbrook, J.D.; et al. RCSB Protein Data Bank: Enabling biomedical research and drug discovery. Protein Sci. 2020, 29, 52–65.
- Sehgal, S.A.; Hammad, M.A.; Tahir, R.A.; Akram, H.N.; Ahmad, F. Current Therapeutic Molecules and Targets in Neurodegenerative Diseases Based on in silico Drug Design. Curr. Neuropharm. 2018, 16, 649–663.
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s disease. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2019; Volume 167, pp. 231–255.
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70.
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554.
- Silva, T.; Reis, J.; Teixeira, J.; Borges, F. Alzheimer’s disease, enzyme targets and drug discovery struggles: From natural products to drug prototypes. Ageing Res. Rev. 2014, 15, 116–145.
- Schliebs, R.; Arendt, T. The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 2011, 221, 555–563.
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335.
- Dvir, H.; Silman, I.; Harel, M.; Rosenberry, T.L.; Sussman, J.L. Acetylcholinesterase: From 3D structure to function. Chem. Biol. Interact. 2010, 187, 10–22.
- Raves, M.L.; Harel, M.; Pang, Y.-P.; Silman, I.; Kozikowski, A.P.; Sussman, J.L. Structure of acetylcholinesterase complexed with the nootropic alkaloid, (–)-huperzine A. Nat. Struct. Mol. Biol. 1997, 4, 57–63.
- da Silva Mesquita, R.; Kyrylchuk, A.; Costa de Oliveira, R.; Costa Sá, I.S.; Coutinho Borges Camargo, G.; Soares Pontes, G.; Moura Araújo da Silva, F.; de Saraiva Nunomura, R.C.; Grafov, A. Alkaloids of Abuta panurensis Eichler: In silico and in vitro study of acetylcholinesterase inhibition, cytotoxic and immunomodulatory activities. PLoS ONE 2020, 15, e0239364.
- Ortiz, J.E.; Garro, A.; Pigni, N.B.; Agüero, M.B.; Roitman, G.; Slanis, A.; Enriz, R.D.; Feresin, G.E.; Bastida, J.; Tapia, A. Cholinesterase-inhibitory effect and in silico analysis of alkaloids from bulbs of Hieronymiella species. Phytomedicine 2018, 39, 66–74.
- Mughal, E.U.; Sadiq, A.; Ashraf, J.; Zafar, M.N.; Sumrra, S.H.; Tariq, R.; Mumtaz, A.; Javid, A.; Khan, B.A.; Ali, A.; et al. Flavonols and 4-thioflavonols as potential acetylcholinesterase and butyrylcholinesterase inhibitors: Synthesis, structure-activity relationship and molecular docking studies. Bioorg. Chem. 2019, 91, 103124.
- Chlebek, J.; Korábečný, J.; Doležal, R.; Štěpánková, Š.; Pérez, D.; Hošťálková, A.; Opletal, L.; Cahlíková, L.; Macáková, K.; Kučera, T.; et al. In Vitro and In Silico Acetylcholinesterase Inhibitory Activity of Thalictricavine and Canadine and Their Predicted Penetration across the Blood-Brain Barrier. Molecules 2019, 24, 1340.
- Kim, J.H.; Thao, N.P.; Han, Y.K.; Lee, Y.S.; Luyen, B.T.T.; Van Oanh, H.; Kim, Y.H.; Yang, S.Y. The insight of in vitro and in silico studies on cholinesterase inhibitors from the roots of Cimicifuga dahurica (Turcz.) Maxim. J. Enzym. Inhib. Med. Chem. 2018, 33, 1174–1180.
- Kiametis, A.S.; Silva, M.A.; Romeiro, L.A.S.; Martins, J.B.L.; Gargano, R. Potential acetylcholinesterase inhibitors: Molecular docking, molecular dynamics, and in silico prediction. J. Mol. Model. 2017, 23, 67.
- Saeedi, M.; Mohtadi-Haghighi, D.; Mirfazli, S.S.; Mahdavi, M.; Hariri, R.; Lotfian, H.; Edraki, N.; Iraji, A.; Firuzi, O.; Akbarzadeh, T. Design and Synthesis of Selective Acetylcholinesterase Inhibitors: Arylisoxazole-Phenylpiperazine Derivatives. Chem. Biodivers. 2019, 16, e1800433.
- Kumar, B.; Kumar, V.; Prashar, V.; Saini, S.; Dwivedi, A.R.; Bajaj, B.; Mehta, D.; Parkash, J.; Kumar, V. Dipropargyl substituted diphenylpyrimidines as dual inhibitors of monoamine oxidase and acetylcholinesterase. Eur. J. Med. Chem. 2019, 177, 221–234.
- Shah, M.S.; Najam-ul-Haq, M.; Shah, H.S.; Farooq Rizvi, S.U.; Iqbal, J. Quinoline containing chalcone derivatives as cholinesterase inhibitors and their in silico modeling studies. Comput. Biol. Chem. 2018, 76, 310–317.
- Ahmad, G.; Rasool, N.; Rizwan, K.; Imran, I.; Zahoor, A.F.; Zubair, M.; Sadiq, A.; Rashid, U. Synthesis, in-vitro cholinesterase inhibition, in-vivo anticonvulsant activity and in-silico exploration of N-(4-methylpyridin-2-yl)thiophene-2-carboxamide analogs. Bioorg. Chem. 2019, 92, 103216.
- Ranjan, A.; Chauhan, A.; Jindal, T. In-silico and in-vitro evaluation of human acetylcholinesterase inhibition by organophosphates. Environ. Toxicol. Pharmacol. 2018, 57, 131–140.
- Castro-Silva, E.S.; Bello, M.; Hernández-Rodríguez, M.; Correa-Basurto, J.; Murillo-Álvarez, J.I.; Rosales-Hernández, M.C.; Muñoz-Ochoa, M. In vitro and in silico evaluation of fucosterol from Sargassum horridum as potential human acetylcholinesterase inhibitor. J. Biomol. Struct. Dyn. 2019, 37, 3259–3268.
- Gurjar, A.S.; Darekar, M.N.; Yeong, K.Y.; Ooi, L. In silico studies, synthesis and pharmacological evaluation to explore multi-targeted approach for imidazole analogues as potential cholinesterase inhibitors with neuroprotective role for Alzheimer’s disease. Bioorg. Med. Chem. 2018, 26, 1511–1522.
- Pereira Rocha, M.; Rodrigues Valadares Campana, P.; de Oliveira Scoaris, D.; de Almeida, V.L.; Dias Lopes, J.C.; Fonseca Silva, A.; Pieters, L.; Gontijo Silva, C. Biological activities of extracts from Aspidosperma subincanum Mart. and in silico prediction for inhibition of acetylcholinesterase. Phyther. Res. 2018, 32, 2021–2033.
- Niu, B.; Zhao, M.; Su, Q.; Zhang, M.; Lv, W.; Chen, Q.; Chen, F.; Chu, D.; Du, D.; Zhang, Y. 2D-SAR and 3D-QSAR analyses for acetylcholinesterase inhibitors. Mol. Divers. 2017, 21, 413–426.
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048.
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33.
- Shearman, E.; Rossi, S.; Szasz, B.; Juranyi, Z.; Fallon, S.; Pomara, N.; Sershen, H.; Lajtha, A. Changes in cerebral neurotransmitters and metabolites induced by acute donepezil and memantine administrations: A microdialysis study. Brain Res. Bull. 2006, 69, 204–213.
- Ivanova, L.; Karelson, M.; Dobchev, D. Identification of Natural Compounds against Neurodegenerative Diseases Using In Silico Techniques. Molecules 2018, 23, 1847.
- Sharma, M.; Mittal, A.; Singh, A.; Jainarayanan, A.K.; Sharma, S.; Paliwal, S. Pharmacophore-driven identification of N-methyl-D-receptor antagonists as potent neuroprotective agents validated using in vivo studies. Biol. Methods Protoc. 2020, 5.
- Waqar, M.; Batool, S. In silico analysis of binding interaction of conantokins with NMDA receptors for potential therapeutic use in Alzheimer’s disease. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 42.
- Hu, S.; Hu, H.; Mak, S.; Cui, G.; Lee, M.; Shan, L.; Wang, Y.; Lin, H.; Zhang, Z.; Han, Y. A Novel Tetramethylpyrazine Derivative Prophylactically Protects against Glutamate-Induced Excitotoxicity in Primary Neurons through the Blockage of N-Methyl-D-aspartate Receptor. Front. Pharmacol. 2018, 9.
- Kumar, S.; Chowdhury, S.; Kumar, S. In silico repurposing of antipsychotic drugs for Alzheimer’s disease. BMC Neurosci. 2017, 18, 76.
- Singh, R.; Ganeshpurkar, A.; Kumar, D.; Kumar, D.; Kumar, A.; Singh, S.K. Identifying potential GluN2B subunit containing N-Methyl-D-aspartate receptor inhibitors: An integrative in silico and molecular modeling approach. J. Biomol. Struct. Dyn. 2020, 38, 2533–2545.

