 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marco Herde | + 1883 word(s) | 1883 | 2021-04-07 11:42:05 | | | |
| 2 | Vicky Zhou | -4 word(s) | 1879 | 2021-04-16 11:31:03 | | |
Video Upload Options
Nucleotides fulfill many essential functions in plants. Compared to non-plant systems, these hydrophilic metabolites have not been adequately investigated in plants, especially the less abundant nucleotide species such as deoxyribonucleotides and modified or damaged nucleotides. Until recently, this was mainly due to a lack of adequate methods for in-depth analysis of nucleotides and nucleosides in plants.
1. Introduction
Nucleotides (NTs), nucleosides (Ns), nucleobases (Nbs), and many derived compounds, for example, nucleotide sugars and nucleotide-containing cofactors, are central metabolites in all organisms (Figure 1). The metabolism of nucleotides in plants and their physiological functions, including those beyond being building blocks of nucleic acids, were recently reviewed [1]. For the investigation of nucleotides in plants, the availability of methods for their comprehensive analysis and robust quantification in plant extracts is pivotal. In this review, we summarize the state of the art for NT and N analysis in plants and also discuss methods used in other organisms that may be applicable for plants. Arguably, the most powerful equipment for metabolite analysis is a chromatographic separation system coupled to an electrospray ionization (ESI) mass spectrometer (MS). This technology is in the focus here. However, our recent study [2] also emphasized the importance of sample preparation for comprehensive NT and N analysis; therefore, we discuss the entire workflow of NT and N analysis in all its aspects—from sample preparation to mass spectrometry—and we comment on how the different steps of the workflow influence each other.
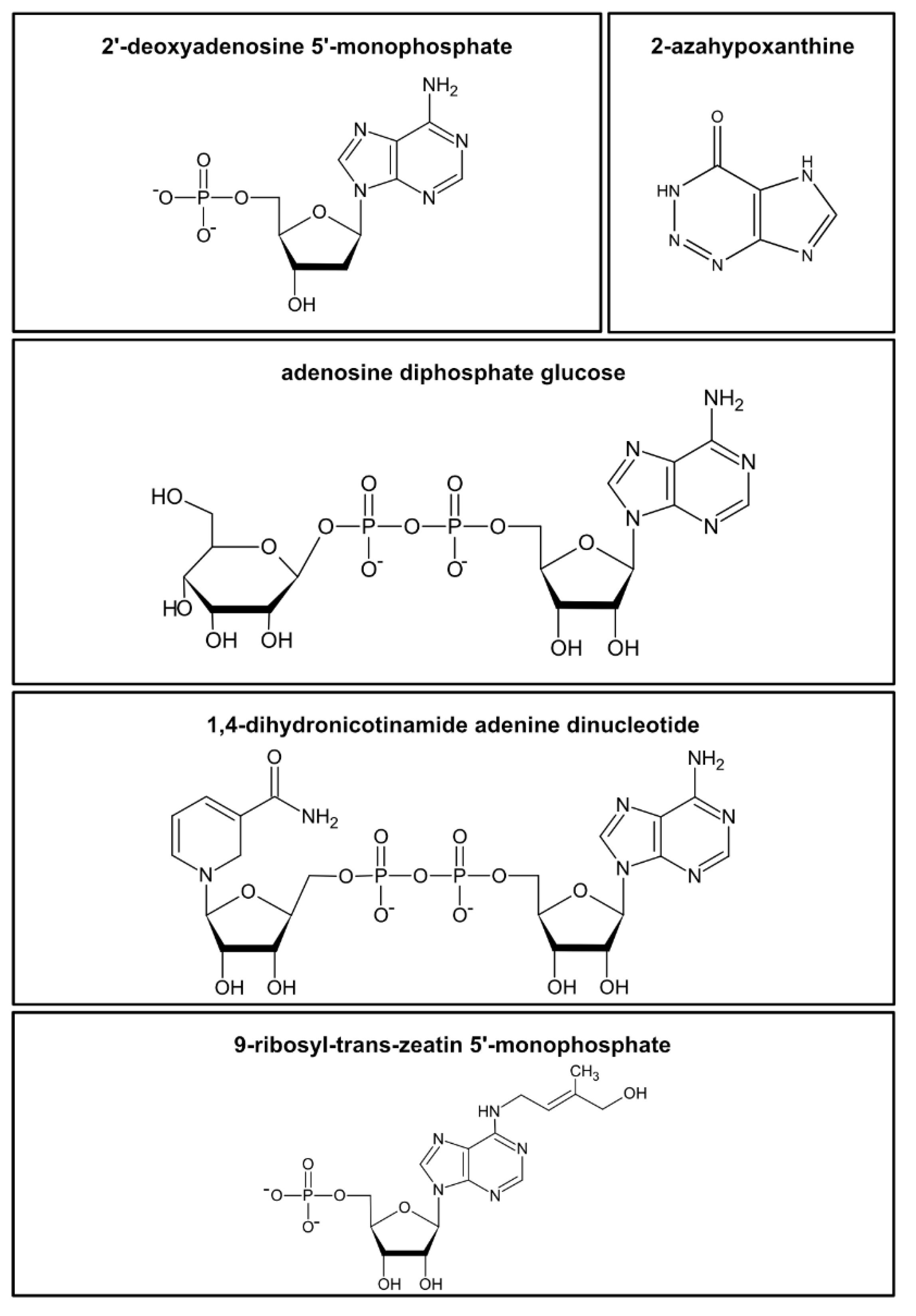
Figure 1. Examples of the diversity of nucleotides and structurally related metabolites in plants. Examples are shown for a nucleotide monophosphate (2′-deoxyadenosine 5′-monophosphate), a “fairy chemical” (2-azahypoxanthine), a nucleotide sugar (adenosine diphosphate glucose), a nucleotide cofactor (1,4-dihydronicotinamide adenine dinucleotide, NAD), and a cytokinine-ribotide (9-ribosyl-trans-zeatin 5′-monophosphate).
The choice of analytical strategies is naturally determined by the physicochemical properties of NTs and Ns. These are polar compounds and the phosphate group(s) of the NTs deprotonate and carry negative charges (pKa values of about 2) [3] within a broad range of pH values. NTs and Ns contain primary, secondary, and tertiary amines, which, especially under more acid conditions, can protonate and acquire a positive charge. In addition, the phosphate groups of nucleotides are negatively charged at a wide range of pH values. [3]. Due to these characteristics, Ns and especially NTs are better soluble in polar (aqueous) solvents.
The concentrations of different NTs vary significantly in plant cells. For example, ribonucleotide triphosphates are about 1000-fold more abundant than deoxyribonucleotide triphosphates [2]. To detect nucleotides of low abundance, additional techniques for enrichment are usually required.
We noticed that methods for nucleotide analysis that are well established in some non-plant systems are not suitable for plants. For example, the simple workflow we applied for the analysis of deoxyribonucleotides (dNTs) in Drosophila [4] failed to work for samples from Arabidopsis thaliana. For the detection of low-abundance nucleotides such as dNTs in phylogenetically diverse plant species, the development of a more complex method was necessary [2]. We can only speculate why the analysis of plants is more complicated, but one contributing factor may be the complexity of the plant matrix which contains a plethora of secondary metabolites [5]. These are probably the reason for the strong ion suppression effects in the ESI source, called matrix effects (ME), which decrease the sensitivity and are usually observed when analyzing plant extracts [2]. Thus, a highly sensitive method for the analysis of the plant NT and N metabolome must include steps to separate these from other interfering metabolites before the chromatographic step. However, if only the more abundant ribonucleosides (rNs) and ribonucleotides (rNTs) are to be monitored, methods for the analysis of the polar metabolome that do not require such extensive sample preparation are sufficient [6][7][8]. For many years, distinct analysis methods for the NT and N metabolome have been evaluated and improved.
2. Extraction of NTs and Ns from Plant Samples
Good solubility of the metabolites in the extraction solvent is crucial for efficient extraction. NTs and Ns are polar and, at least in the case of NTs, also charged, requiring a polar solvent for efficient extraction. Often the extractant is aqueous [2][9], but water/acetonitrile mixtures are in use as well [10][11][12]. The solubility of the analytes in the solvent should be considered when choosing the extractant volume in relation to the sample amount, especially when less soluble Nbs [3] are extracted. If solubility is limiting, re-extraction of a pellet will result again in high analyte concentration in the supernatant. The solubility is also dependent on the pH of the extractant. For the extraction of polar metabolites, a 1:10 ratio of fresh material to solvent is recommended, whereas a ratio of 1:100 is recommended for lyophilized material [13].
Extraction and all following steps should be designed to stabilize the analytes. This is essential for NTs, since they may lose a phosphate moiety, for example, during chromatography [14]. One study found that acidification of the extractant can substantially improve the stability of NTs [10]. Our recent work confirmed that, at least for the duration of the extraction, NTs and Ns are acid-stable with good recoveries [2]. Although more studies have used acidic conditions for NT extraction, there are protocols for the determination of rNTs in Nicotiana tabacum, Arabidopsis thaliana, and Solanum lycopersicum using quenching with a high pH, which also led to excellent recoveries [15]. At high pH, NTs were reported to be stable even when stored for 3 days at room temperature [15]. In general, frozen nucleotide solutions are stable for a prolonged time if the acid from the quenching step is removed or neutralized [16]. With methods that directly use clarified crude extracts for MS analysis, extraction may result in the release of metabolites that cause ion suppression and, thus, decrease the sensitivity in mass spectrometry (matrix effect) [10]. A further concern is that the concentrations of NTs and Ns may be overestimated if the extraction procedure can potentially release them from other metabolites. The most obvious example is the hydrolytic release of phosphate groups from nucleotide triphosphates (NTPs), reducing NTP concentrations and leading to an overestimation of nucleotide diphosphates (NDPs), nucleotide monophosphates (NMPs), or Ns. Another example is the release of uridine diphosphate (UDP) from UDP-glucose, since the hydrolysis of this compound was observed under mild acidic conditions and at an elevated temperature in nucleotide sugar analysis [17].
Adding a non-polar solvent immiscible with water to the extraction will result in the partitioning of Ns and NTs to the polar/aqueous phase and of non-polar metabolites to the non-polar phase. Separation and individual processing of both phases results in a coarse fractionation of metabolites which is used in studies simultaneously reporting polar and non-polar metabolites [18]. As a non-polar solvent immiscible with water, methy-tert-butyl-ether (MTBE) is a less toxic alternative to fluorocarbons such as 1,1,2-trichlor-1,2,2-trifluorethan used previously [19] and is, therefore, more environmentally friendly and safer to handle [18]. Moreover, dichloromethane and water-saturated diethyl ether have been used as less toxic alternatives [2][16]. Hexane or chloroform was employed in some studies for the removal of phospholipids by LLE [20][21]. The addition of polyvinylpolypyrrolidone can remove phenolic compounds that cannot be eliminated by other means and can interfere with the detection of NTs [22][23].
The rNTs and rNs, but not their dNT and deoxyribonucleoside (dN) counterparts, have two neighboring hydroxyl groups (cis-diol) in the sugar moiety. This feature can be used either for the extraction of rNTs and rNs (see next chapter) or for their selective degradation by periodate and methylamine-driven β-elimination [22][24][25][26]. Selective degradation of the more abundant rNTs and rNs can enhance the detection of dNs and dNTs by reducing the specific NT/N background. This can be explained, for example, by less competition for binding to the resin during solid-phase extraction or simply by less ion suppression in the ESI source. Selective cis-diol degradation may also be an improvement for detecting NTs and Ns with modified sugars such as 2′-O-methylguanosine, because these will be protected from degradation. When using the selective degradation protocol, it has been recommended to first determine the recovery rates of the metabolites in focus, because, even though dNTs are generally not degraded, deoxyguanosine triphosphate (dGTP), for example, can react with periodate [27].
3. Outlook
Although the comprehensive quantification of Ns and NTs is now possible in samples from a variety of plants and algae, many challenges still remain. While current workflows can detect the relatively low abundant dNs and dNTs in plants [2][22], reports about the detection of damaged or modified NTs which are probably even less abundant are still scarce [28][29]. Since such rare NTs are more frequently observed in non-plant systems [30][31], it is tempting to speculate that it is the complexity of the plant matrix which hampers their detection in plants. It will be necessary to boost the sensitivity to also detect these rare NTs, which may be achieved in part by further reducing the complexity of the matrix, for example, by eliminating the more abundant rNTs [22][26]. Additionally, up-scaling of the sample amount in combination with enrichment techniques and derivatization protocols might be part of a solution.
Robust sample preparation coupled with a high sensitivity of detection will improve the analysis of NTs and Ns in cases where only little starting material is available, for example, when only a certain plant tissue is investigated. It will also be feasible to analyze plants with a particularly complex metabolome and correspondingly complex matrix (e.g., in Viscum album [32]).
Some rNTs and rNs but not the dNs and dNTs or the modified NTs were previously reported in studies aiming at the description of the whole (polar) metabolome [6][7]. Currently, there is a tradeoff between the depth of NT analysis on one side and the ability to comprehensively describe the entire metabolome on the other side. Coupling our sample preparation protocol for NT and N analysis [2] with protocols for the simultaneous analysis of amino acids, phytohormones, and lipids [18] may help to overcome this problem. Although the sample preparation would be more time-consuming, the preparation of a single sample may be sufficient for a more comprehensive in-depth analysis of the metabolome. Potentially, several groups of non-polar but also polar and less abundant metabolites such as phytohormones, (d)NTs, and (d)Ns could be quantified from such samples. An alternative approach uses a combination of reverse-phase and HILIC chromatography to monitor lipids and polar metabolites in one analytical run [33].
A major drawback of LC–MS techniques is that they are unable to monitor NTs in living cells and cannot assess NT concentrations in particular cells within complex tissues or in subcellular compartments. For some abundant NTs such as ATP and nicotinamide adenine dinucleotide, molecular probes were developed to monitor the in vivo concentrations within Arabidopsis thaliana cells and tissues [34][35]. Although such probes are currently only available for very few metabolites and are less sensitive than LC–MS (a concentration of ~160 µM can be detected with the ATP sensor, compared to a sensitivity in the picomolar range for LC–MS), these techniques can complement data obtained by LC–MS. Techniques to fractionate [36][37] or isolate subcellular compartments [38] in combination with metabolite analysis via LC–MS may prove useful for the investigation of the subcellular NT and N pools in plant cells. In the future, such fractionation and isolation techniques must gain in resolution. Especially for organelle isolation techniques, quenching strategies must be devised to effectively prevent alterations to the metabolome during organelle preparation.
References
- Witte, C.-P.; Herde, M. Nucleotide metabolism in plants. Plant Physiol. 2020, 182, 63–78.
- Straube, H.; Niehaus, M.; Zwittian, S.; Witte, C.-P.; Herde, M. Enhanced nucleotide analysis enables the quantification of deoxynucleotides in plants and algae revealing connections between nucleoside and deoxynucleoside metabolism. Plant Cell 2020.
- Hodgson, D.R.W. Physicochemical Aspects of Aqueous and Nonaqueous Approaches to the Preparation of Nucleosides, Nucleotides and Phosphate Ester Mimics. Adv. Phys. Org. 2017, 51, 187–219.
- Liu, B.; Winkler, F.; Herde, M.; Witte, C.-P.; Grosshans, J. A Link between deoxyribonucleotide metabolites and embryonic cell-cycle control. Curr. Biol. 2019, 29, 1187.
- Bielski, R.L. The problem of halting enzyme action when extracting plant tissues. Anal. Biochem. 1964, 9, 431–442.
- Creydt, M.; Fischer, M. Plant metabolomics: Maximizing metabolome coverage by optimizing mobile phase additives for nontargeted mass spectrometry in positive and negative electrospray ionization mode. Anal. Chem. 2017, 89, 10474–10486.
- Rolletschek, H.; Melkus, G.; Grafahrend-Belau, E.; Fuchs, J.; Heinzel, N.; Schreiber, F.; Jakob, P.M.; Borisjuk, L. Combined noninvasive imaging and modeling approaches reveal metabolic compartmentation in the barley endosperm. Plant Cell 2011, 23, 3041–3054.
- De Souza, A.P.; Cocuron, J.-C.; Garcia, A.C.; Alonso, A.P.; Buckeridge, M.S. Changes in whole-plant metabolism during the grain-filling stage in sorghum grown under elevated CO2 and drought. Plant Physiol. 2015, 169, 1755–1765.
- Baccolini, C.; Witte, C.-P. AMP and GMP catabolism in arabidopsis converge on xanthosine, which is degraded by a nucleoside hydrolase heterocomplex. Plant Cell 2019, 31, 734–751.
- Rabinowitz, J.D.; Kimball, E. Acidic acetonitrile for cellular metabolome extraction from Escherichia coli. Anal. Chem. 2007, 79, 6167–6173.
- Au, J.L.; Su, M.H.; Wientjes, M.G. Extraction of intracellular nucleosides and nucleotides with acetonitrile. Clin. Chem. 1989, 35, 48–51.
- Chen, H.; Zhang, B.; Hicks, L.M.; Xiong, L. A nucleotide metabolite controls stress-responsive gene expression and plant development. PLoS ONE 2011, 6.
- Liu, Z.; Rochfort, S. Recent progress in polar metabolite quantification in plants using liquid chromatography-mass spectrometry. J. Integr. Plant Biol. 2014, 56, 816–825.
- Zbornikova, E.; Knejzlik, Z.; Hauryliuk, V.; Krasny, L.; Rejman, D. Analysis of nucleotide pools in bacteria using HPLC-MS in HILIC mode. Talanta 2019, 205.
- Riondet, C.; Morel, S.; Alcaraz, G. Determination of total ribonucleotide pool in plant materials by high-pH anion-exchange high-performance liquid chromatography following extraction with potassium hydroxide. J. Chromatogr. A 2005, 1077, 120–127.
- Brown, P.R. Stability of nucleotide solutions on storage as determined by high-pressure liquid chromatography. Anal. Biochem. 1971, 43, 305–306.
- Barnes, J.; Tian, L.; Loftis, J.; Hiznay, J.; Comhair, S.; Lauer, M.; Dweik, R. Isolation and analysis of sugar nucleotides using solid phase extraction and fluorophore assisted carbohydrate electrophoresis. MethodsX 2016, 3, 251–260.
- Salem, M.A.; Yoshida, T.; de Souza, L.P.; Alseekh, S.; Bajdzienko, K.; Fernie, A.R.; Giavalisco, P. An improved extraction method enables the comprehensive analysis of lipids, proteins, metabolites and phytohormones from a single sample of leaf tissue under water-deficit stress. Plant J. 2020, 103, 1614–1632.
- Khym, J.X. An analytical system for rapid separation of tissue nucleotides at low pressures on conventional anion exchangers. Clin. Chem. 1975, 21, 1245–1252.
- Soga, T.; Ishikawa, T.; Igarashi, S.; Sugawara, K.; Kakazu, Y.; Tomita, M. Analysis of nucleotides by pressure-assisted capillary electrophoresis-mass spectrometry using silanol mask technique. J. Chromatogr. A 2007, 1159, 125–133.
- Cordell, R.L.; Hill, S.J.; Ortori, C.A.; Barrett, D.A. Quantitative profiling of nucleotides and related phosphate-containing metabolites in cultured mammalian cells by liquid chromatography tandem electrospray mass spectrometry. J. Chromatogr. B 2008, 871, 115–124.
- Dutta, I.; Dutta, P.K.; Smith, D.W.; O’Donovan, G.A. High-performance liquid-chromatography of deoxynucleoside diphosphates and triphosphates in tomato roots. J. Chromatogr. A 1991, 536, 237–243.
- Nieman, R.H.; Pap, D.L.; Clark, R.A. Rapid purification of plant nucleotide extracts with xad-2,polyvinyl-polypyrrolidone and charcoal. J. Chromatogr. A 1978, 161, 137–146.
- Tanaka, K.; Yoshioka, A.; Tanaka, S.; Wataya, Y. An improved method for the quantitative determination deoxyribonucleoside triphosphates in cell-extracts. Anal. Biochem. 1984, 139, 35–41.
- Uziel, M. Periodate oxidation and amine-catalyzed elimination of terminal nucleoside from adenylate or ribonucleic-acid products of overoxidation. Biochemistry 1973, 12, 938–941.
- Odmark, G.; Kihlman, B.A. Effects of chromosome-breaking purine derivates on nucleic acid synthesis and on levels of adenosine 5′-triphosphate and deoxyadenosine 5′-triphosphate in bean root tips. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1965, 2, 274–286.
- Hennere, G.; Becher, F.; Pruvost, A.; Goujard, C.; Grassi, J.; Benech, H. Liquid chromatography-tandem mass spectrometry assays for intracellular deoxyribonucleotide triphosphate competitors of nucleoside antiretrovirals. J. Chromatogr. B 2003, 789, 273–281.
- Chen, M.; Witte, C.-P. A kinase and a glycosylase catabolize pseudouridine in the peroxisome to prevent toxic pseudouridine monophosphate accumulation. Plant Cell 2020, 32, 722–739.
- Chen, M.; Urs, M.J.; Sanchez-Gonzalez, I.; Olayioye, M.A.; Herde, M.; Witte, C.-P. m(6)A RNA degradation products are catabolized by an evolutionarily conserved N-6-Methyl-AMP deaminase in plant and mammalian cells. Plant Cell 2018, 30, 1511–1522.
- Jiang, H.-P.; Xiong, J.; Liu, F.-L.; Ma, C.-J.; Tang, X.-L.; Yuan, B.-F.; Feng, Y.-Q. Modified nucleoside triphosphates exist in mammals. Chem. Sci. 2018, 9, 4160–4167.
- Galperin, M.Y.; Moroz, O.V.; Wilson, K.S.; Murzin, A.G. House cleaning, a part of good housekeeping. Mol. Microbiol. 2006, 59, 5–19.
- Senkler, J.; Rugen, N.; Eubel, H.; Hegermann, J.; Braun, H.-P. Absence of complex I implicates rearrangement of the respiratory chain in european mistletoe. Curr. Biol. 2018, 28, 1606.
- Schwaiger, M.; Schoeny, H.; El Abiead, Y.; Hermann, G.; Rampler, E.; Koellensperger, G. Merging metabolomics and lipidomics into one analytical run. Analyst 2019, 144, 220–229.
- Voon, C.P.; Guan, X.; Sun, Y.; Sahu, A.; Chan, M.N.; Gardestrom, P.; Wagner, S.; Fuchs, P.; Nietzel, T.; Versaw, W.K.; et al. ATP compartmentation in plastids and cytosol of Arabidopsis thaliana revealed by fluorescent protein sensing. Proc. Natl. Acad. Sci. USA 2018, 115, E10778–E10787.
- Steinbeck, J.; Fuchs, P.; Negroni, Y.L.; Elsaesser, M.; Lichtenauer, S.; Stockdreher, Y.; Feitosa-Araujo, E.; Kroll, J.B.; Niemeier, J.-O.; Humberg, C.; et al. In Vivo NADH/NAD+ Biosensing Reveals the Dynamics of Cytosolic Redox Metabolism in Plants. Plant Cell 2020, 32, 3324–3345.
- Arrivault, S.; Guenther, M.; Florian, A.; Encke, B.; Feil, R.; Vosloh, D.; Lunn, J.E.; Sulpice, R.; Fernie, A.R.; Stitt, M. Dissecting the subcellular compartmentation of proteins and metabolites in arabidopsis leaves using non-aqueous fractionation. Mol. Cell. Proteom. 2014, 13, 2246–2259.
- Fuertauer, L.; Kuestner, L.; Weckwerth, W.; Heyer, A.G.; Naegele, T. Resolving subcellular plant metabolism. Plant J. 2019, 100, 438–455.
- Niehaus, M.; Straube, H.; Kuenzler, P.; Rugen, N.; Hegermann, J.; Giavalisco, P.; Eubel, H.; Witte, C.-P.; Herde, M. Rapid affinity purification of tagged plant mitochondria (Mito-AP) for metabolome and proteome analyses. Plant Physiol. 2020, 182, 1194–1210.


