 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Feng Wan | + 2149 word(s) | 2149 | 2021-04-15 05:53:49 | | | |
| 2 | Peter Tang | Meta information modification | 2149 | 2021-04-16 03:57:09 | | |
Video Upload Options
Diffuse intrinsic pontine gliomas are malignant brain tumors which arise from the pons in children. These tumors are incurable and nearly all the patients die within a year after diagnosis.
1. Introduction
Diffuse intrinsic pontine glioma (DIPG), a high-grade glioma that arises in the pons is predominantly seen in children. This central nervous system (CNS) malignancy represents the leading cause of brain-tumor related death [1]. Ionizing radiation extends overall survival to a median of 11 months [2]. Numerous clinical trials have failed to identify effective agents, or therapeutic combinations against DIPG [3]. A key to identifying improved treatments is to enhance our biological understanding of this tumor.
DIPG molecular signature has been profiled over the last decade. Landmark studies from multiple groups identified an epigenetic oncogenic histone H3K27M mutation in ~80% of DIPGs [4][5][6]. This mutation was defined as a new entity labeled “diffuse midline glioma H3K27M-mutant” in the 2016 World Health Organiztion (WHO) tumor classification [7]. A number of molecular aberrations, which are potential targets for treatment have been identified. They include RB phosphorylation (~30%) [8], p53, Wee1, STAT3, PPM1D (9–23%) and EGFRvIII overexpression [9][10][11][12][13], platelet-derived growth factor receptor A (PDGFRA) (30%) and MET (26%) amplification [8][14], and ACVR1 (21%) [15][16][17][18], PI3K-mTOR or -MAPK (62%) activation [8]. These aberrations are either independent or concurrent with H3K27M mutations in DIPGs. More recently, multiple pathways, such as Notch signaling [19], glycolysis and tricarboxylic acid (TCA) metabolic pathways [20], Wilms’ tumor protein (WT1) overexpression [21][22] and FGFR2-VPS35 fusion [23] were delineated. These pathways are also potential therapeutic targets. Altogether, these abnormalities indicate the molecular complexity of DIPGs. Thus, to identify effective targeting therapeutics or combinations of such, reliable and personalized animal models are desirable for precise preclinical evaluation prior to clinical trials.
Various animal models have been developed for the identification of potential therapeutics against DIPGs. One reason for this development was that historically, surgical biopsies for DIPGs were not performed due to the critical nature of the brainstem, limitations of surgical techniques and histological heterogeneity within the tumor [24]. Multiple murine gliomas induced with various carcinogens were transplanted, so called “allografts”, into the brainstem as models for DIPGs [25]. The results from these models, however, failed to translate into clinical trials due to differences in tumorigenesis mechanisms between these murine gliomas and DIPGs in children. Recently, several groups have safely performed biopsies and obtained tumor specimens from DIPG patients [26]. Moreover, adequate autopsy DIPG specimens have been obtained for experimental purposes. With availability of these invaluable specimens, more accurate patient-derived orthotopic xenograft (PDOX) DIPG mouse models were and continue to be generated for pre-clinical therapeutic testing [27][28][29]. More importantly, these specimens helped generate multiple cell-based models, which have provided significant insights into genetic and epigenetic alterations driving DIPGs. These insights, in conjunction with current cutting-edge gene editing techniques, have allowed and prompted scientists to generate robust genetically engineered mouse models (GEMMs), to understand DIPG tumorigenesis and to provide precise molecular models for pre-clinical testing. One caveat is that the majority of xenograft models are produced in immunodeficient athymic or NOD-SCID gamma (NSG) mice, which lack normal immunity. Consequently, these mouse models do not mimic the human tumor microenvironment including infiltration by immune cells. Recent work has shown that immune cells discovered in pediatric DIPG tumor specimens are distinct and differ from those in adult glioblastomas [30]. Generation of humanized DIPG mouse models is urgently needed to explore these differences, an understanding of which will accelerate therapeutic discovery.
2. Molecular Characteristics of DIPG
Diffuse intrinsic pontine glioma (DIPG) was identified in 1926 [31]. For decades, its biological behavior was thought to be similar to adult malignant gliomas, thus therapeutic regimens for adult tumors were copied in children. These treatments failed to improve patient outcomes [3], which led researchers to ask if pediatric malignant gliomas including DIPGs fundamentally differ from adult gliomas.
Recently several groups have developed safe and feasible methods to collect biopsy tissue samples from pediatric DIPG patients with minimum mortality [26][32], though routine DIPG biopsy continues to be debated. Furthermore, autopsy samples can be acquired for either direct molecular profiling or development of patient-derived primary cell lines and/or xenografts to gain insight into tumor driving mechanisms [33]. These tissues sources, as well as high-throughput genetic technologies enabled researchers to acquire data, which provided insight into DIPG molecular signatures. For instance, recurrent amplifications of PDGFRA, MET and retinoblastoma protein (RB) are unique for pediatric DIPG [8][34]. These findings prove that DIPG is molecularly distinct from adult malignant gliomas. Other groups using high-throughput genetic sequencing technologies have identified that ~80% DIPGs contain somatic point heterozygous H3.1K27M or H3.3K27M mutations [4][5][6], recognized to be major oncogenic drivers in these tumors [6][35][36].
DIPGs with H3.1K27M or H3.3K27M mutations, coded by HIST1H3B, HIST1H3C and H3F3A, respectively [37] have different clinical manifestations and gene expression profiles. H3.1K27M tumors are usually restricted to the pons, and show a mesenchymal-like phenotype, with an overall survival of 15 months. Tumors with H3.3K27 mutation are found in the pons and other midline locations such as the thalamus, with an overall survival of 9 months [38][39][40]. These tumors display an oligodendroglial-like phenotype and are more resistant to radiation therapy [40]. Both H3.1K27M and H3.3K27M mutations primarily influence the epigenome and are required for DIPG tumorigenesis and maintenance [41][42]. These mutations predominantly reduce genome-wide levels of repressive H3K27me3 [37][43][44] through inactivation of Polycomb Repressive Complex 2 (PRC2) [45]. Further studies have confirmed that H3K27M suppresses PRC2 through tight binding to EZH2, a core subunit of PRC2 [46]. H3K27me3 levels are differentially associated with H3.1K27M or H3.3K27M mutations, with H3.3K27M epigenome maintaining a certain amount of H3K27me3, while H3.1K27M almost completely excludes genomic-wide H3K27me3 [47]. Moreover, H3K27M DIPG shows slightly elevated H3K27ac [45][48][49], which colocalizes with H3K27M mutations at enhancer or promoter areas [45]. Multiple pre-clinical therapeutic evaluations have shown that inhibition of histone deacetylases is effective and has survival benefits in DIPG xenograft animal models [50], indicating therapeutic potential targeting H3K27ac in H3K27M mutant DIPGs. Interestingly, global H3K4me3 levels are relatively stable regardless of histone H3 mutation [42], however, promoter H3K4me3 level is higher in H3K27M mutants than in WT tumors, at specific gene loci including Lin28b, a marker for neural stem cells [51]. More recently, H3K36me2 and H4K16ac were identified as important histone marks in DIPGs [52]. These findings indicated complicated crosstalk among posttranslational histone modifications, the underlining mechanisms are still under investigation.
In addition to epigenomic alterations in H3K27M mutant DIPG, numerous aberrations of gene expression, DNA copy number variations and signal pathways have been uncovered. These may occur concurrently or independently with H3K27M mutation. p38 MAPK is activated in both H3.3K27M and H3.1K27M cultured cells with H3.1K27M tumor cells more sensitive to p38 MAPK inhibition [49]. WNT [49][53], mTOR [53][54] and RTK-RAS-PI3K signaling pathways [53][55] were also active in both tumor subtypes. Interestingly, H3.1K27M and H3.3K27M DIPGs have their own associated mutations as summarized in Figure 1.
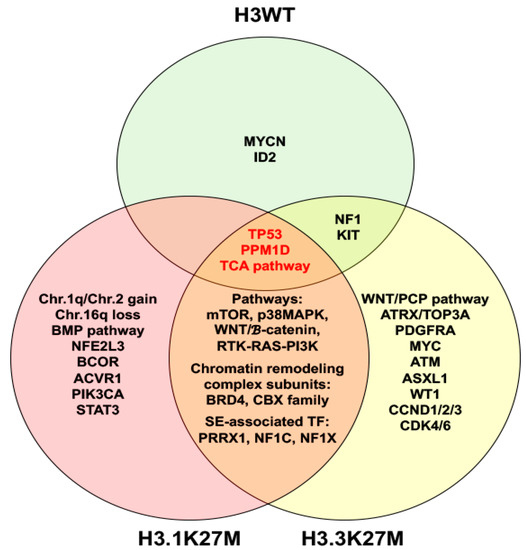
Figure 1. Venn diagram illustrates molecular characteristics of histone H3 wild type (H3WT), H3.1K27M and H3.3K27M diffuse intrinsic pontine gliomas. Abbreviation: TCA, tricarboxylic acid.
The diversity in genetics, chromatin landscape and metabolic reprogramming of DIPGs clearly shows that individualized therapeutics will be critical for effective treatments. To this end, developing personalized animal models for pre-clinical assessment is an important step to identify and determine the best therapeutic agents.
3. Humanized Mouse Models for DIPG Precision Medicine
Recently, serval studies have shown that the tumor immune microenvironment (TIME) has critical roles in DIPG: (i) Tumor-infiltrating cells (TILs) including Treg, CD4 T cells, NK, B cells, monocytes and eosinophils have been identified in H3K27M mutant DIPGs [56]; (ii) Expression of indoleamine 2,3 dioxygenase 1 (IDO1), an immunosuppressive enzyme that metabolizes tryptophan, is low in cultured DIPG cells. However, in vitro induction of IDO1 with IFNγ showed potential therapeutic value [57]; (iii) the disialoganglioside GD2 is highly expressed in patient-derived H3K27M mutant glioma cell cultures. Anti-GD2 CAR T cells incorporating a 4-1BBz costimulatory domain demonstrated robust antigen-dependent cytokine generation and killed DIPG cells in vitro [58] and (iv) a humanized anti-CD47 antibody, Hu5F9-G4, demonstrated therapeutic efficacy [59].
These cutting-edge findings require testing using reliable animal models prior to translation into clinical trials. For many years, chimpanzees were used to bridge the gap between rodent models and humans. However, the biomedical use of chimpanzees is prohibited in Europe and the United States. Therefore, to overcome the limitations of translating laboratory rodent discoveries into clinical applications, development of mouse models that closely recapitulate human biological systems, labeled as “humanized” mice is critical for pre-clinical investigation. In this section, we will discuss recent progress in the development and potential use of humanized DIPG mouse models.
Humanized mice are defined as immunodeficient mice engrafted with human tissues [60], which include PDX or PDOX models. Discovery of nude mice in the 1960s and severe combined immunodeficient (SCID) mice in the 1980s [61] were key advances for xenografts. Following these models, non-obese diabetic (NOD)/SCID and NOD/SCID /β2mnull and NOD/Rag1nullPfpnull mice were subsequently developed from NOD/SCID and NOD/Rag1nullmice [61], which contribute to humanized mouse generation. Another landmark advance in the generation of humanized mice was the generation of NOD/SCID/γcnull mice and Rag1/2nullγcnull mice through introducing IL2ra into NOD/SCID and RAG1/2−/− mice in the 2000s [61]. These mice show multiple immunodeficiencies including impaired T, B and nature killer (NK) cells, and reduced macrophages and dendritic cell immune function, which show a high rate of human cell including DIPG engraftment. More recently, several humanized mouse models were used to test novel potential therapeutics for DIPG. NRG (NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl/SzJ) mice, which had DIPG cells implanted in the pons, were used to test therapeutic efficacy of a DNA-damaging reagent 6-thio-2′deoxyguanosine (6-thio-dG). The results demonstrated promising therapeutic efficacy [62]. NOD-SCIDγ (NSG) mice were used to test anti-CD47 antibody Hu5F9-G4 through orthotopic injection of several DIPG cells. The results showed intraperitoneal treatment with this antibody significantly reduced tumor growth and showed significant survival benefit [59]. NSG NOD-SCID IL2rg−/− (NSG) mice were used for testing anti-GD2 CAR-T cell immunotherapy [58], with the results leading to an active clinical trial (NCT04196413).
Humanized mice are also defined as immunodeficient mice engrafted with hemato-poietic cells [60][61]. There are several strategies for establishment of these humanized mouse models (Figure 2). The first one is humanized mice receiving human peripheral blood mononuclear cells (PBMCs) engrafted to establish the Hu-PBL-SCID model. This model is suitable for short-term research and investigation of the relationship between immune function of lymphocytes in peripheral blood and tumor biological behavior [63]. The second strategy is the transfer of human hematopoietic stem cells (HSCs) into mice with the IL-2rγnull mutation to develop the Hu-SRC-SCID model [64]. HSCs can be obtained from granulocyte colony-stimulating factor-mobilized PBMCs, adult bone marrow, fetal liver and umbilical cord [65]. This model also supports engraftment of complete human immune system through injection of CD34+ HSCs (Hu-CD34+ model), which is appropriate for investigating tumor growth and immune system development [66]. The third model is a bone marrow/liver/thymus (BLT) model which is developed via transplanting human fetal liver and thymus under the kidney capsule. More recently, a novel and revolutionary humanized mouse model NOD-SCID IL2rgnull SCF/GM-CSF/IL3 strain engrafted with human thymus, liver, and hematopoietic stem cells (termed Bone marrow, Liver, Thymus [BLT]) (NSG-SGM3-BLT) was used to develop an orthotopic model through injection of SF8628, a H3.3K27M mutant cell in the pons, to test IDO1 induction by CD4+ and CD8+ T cells. The results showed that these T cells directly increase IDO1 expression in intracranial DIPG tumor and are thus a promising adjuvant immunotherapy [57].
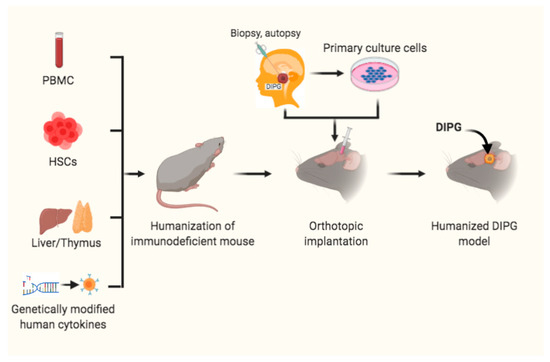
Figure 2. Humanized mouse models for DIPG. Immunodeficient mice received peripheral blood mononuclear cells (PBMCs), or hematopoietic stem cells (HSCs) and transplantation of liver/thymus, or genetically modified human cytokines prior to orthotopic intracranial inoculation of DIPG cells, from patient biopsy or autopsy specimens or primary cultured cells, into the pons to develop humanized DIPG models.
In addition, mice transgenically engineered to express human genes are also humanized mice. One example is MISTRG mice, in which seven genes including M-CSFh/h IL-3/GM-CSFh/h SIRPah/h TPOh/h RAG2−/− IL2Rg−/− were knocked into mouse genomic loci [67]. MISTRG mice are humanized with high immunodeficiency, which prevents immune rejection of the human grafts. These mice are a robust tool to investigate engrafted tumors and innate immune response, which is potentially useful for the development of humanized DIPG models.
Altogether, these advanced humanized mouse models provide a more realistic human tumor immune microenvironment with potential for better drug response and prediction for clinical trials and will help to identify effective therapeutic regimens for DIPG.
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-oncology 2019, 21, v1–v100.
- Cooney, T.; Lane, A.; Bartels, U.; Bouffet, E.; Goldman, S.; Leary, S.E.S.; Foreman, N.K.; Packer, R.J.; Broniscer, A.; E Minturn, J.; et al. Contemporary survival endpoints: An International Diffuse Intrinsic Pontine Glioma Registry study. Neuro-Oncology 2017, 19, 1279–1280.
- Hargrave, D.; Bartels, U.; Bouffet, E. Diffuse brainstem glioma in children: Critical review of clinical trials. Lancet Oncol. 2006, 7, 241–248.
- Khuong-Quang, D.-A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.-Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447.
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253.
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Khuong-Quang, D.-A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231.
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820.
- Paugh, B.S.; Broniscer, A.; Qu, C.; Miller, C.P.; Zhang, J.; Tatevossian, R.G.; Olson, J.M.; Geyer, J.R.; Chi, S.N.; Da Silva, N.S.; et al. Genome-Wide Analyses Identify Recurrent Amplifications of Receptor Tyrosine Kinases and Cell-Cycle Regulatory Genes in Diffuse Intrinsic Pontine Glioma. J. Clin. Oncol. 2011, 29, 3999–4006.
- Park, J.; Lee, W.; Yun, S.; Kim, S.P.; Kim, K.H.; Kim, J.; Kim, S.; Wang, K.; Lee, J.Y. STAT3 is a key molecule in the oncogenic behavior of diffuse intrinsic pontine glioma. Oncol. Lett. 2020, 20, 1989–1998.
- Wang, Z.; Xu, C.; Diplas, B.H.; Moure, C.J.; Chen, C.-P.J.; Chen, L.H.; Du, C.; Zhu, H.; Greer, P.K.; Zhang, L.; et al. Targeting Mutant PPM1D Sensitizes Diffuse Intrinsic Pontine Glioma Cells to the PARP Inhibitor Olaparib. Mol. Cancer Res. 2020, 18, 968–980.
- Li, G.; Mitra, S.S.; Monje, M.; Henrich, K.N.; Bangs, C.D.; Nitta, R.T.; Wong, A.J. Expression of epidermal growth factor variant III (EGFRvIII) in pediatric diffuse intrinsic pontine gliomas. J. Neuro-Oncol. 2012, 108, 395–402.
- Solomon, D.A.; Wood, M.D.; Tihan, T.; Bollen, A.W.; Gupta, N.; Phillips, J.J.J.; Perry, A. Diffuse Midline Gliomas with Histone H3-K27M Mutation: A Series of 47 Cases Assessing the Spectrum of Morphologic Variation and Associated Genetic Alterations. Brain Pathol. 2016, 26, 569–580.
- Mueller, S.; Hashizume, R.; Yang, X.; Kolkowitz, I.; Olow, A.K.; Phillips, J.; Smirnov, I.; Tom, M.W.; Prados, M.D.; James, C.D.; et al. Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro-Oncology 2013, 16, 352–360.
- Paugh, B.S.; Zhu, X.; Qu, C.; Endersby, R.; Diaz, A.K.; Zhang, J.; Bax, D.A.; Carvalho, D.; Reis, R.M.; Onar-Thomas, A.; et al. Novel Oncogenic PDGFRA Mutations in Pediatric High-Grade Gliomas. Cancer Res. 2013, 73, 6219–6229.
- Fortin, J.; Tian, R.; Zarrabi, I.; Hill, G.; Williams, E.; Sanchez-Duffhues, G.; Thorikay, M.; Ramachandran, P.; Siddaway, R.; Wong, J.F.; et al. Mutant ACVR1 Arrests Glial Cell Differentiation to Drive Tumorigenesis in Pediatric Gliomas. Cancer Cell 2020, 37, 308–323.e12.
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.S.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461.
- Hoeman, C.M.; Cordero, F.J.; Hu, G.; Misuraca, K.; Romero, M.M.; Cardona, H.J.; Nazarian, J.; Hashizume, R.; McLendon, R.; Yu, P.; et al. ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nat. Commun. 2019, 10, 1023.
- Cao, H.; Jin, M.; Gao, M.; Zhou, H.; Tao, Y.J.; Skolnick, J. Differential kinase activity of ACVR1 G328V and R206H mutations with implications to possible TβRI cross-talk in diffuse intrinsic pontine glioma. Sci. Rep. 2020, 10, 6140.
- Taylor, I.C.; Hütt-Cabezas, M.; Brandt, W.D.; Kambhampati, M.; Nazarian, J.; Chang, H.T.; Warren, K.E.; Eberhart, C.G.; Raabe, E.H. Disrupting NOTCH Slows Diffuse Intrinsic Pontine Glioma Growth, Enhances Radiation Sensitivity, and Shows Combinatorial Efficacy with Bromodomain Inhibition. J. Neuropathol. Exp. Neurol. 2015, 74, 778–790.
- Chung, C.; Sweha, S.R.; Pratt, D.; Tamrazi, B.; Panwalkar, P.; Banda, A.; Bayliss, J.; Hawes, D.; Yang, F.; Lee, H.-J.; et al. Integrated Metabolic and Epigenomic Reprograming by H3K27M Mutations in Diffuse Intrinsic Pontine Gliomas. Cancer Cell 2020, 38, 334–349.e9.
- Lee, S.; Kambhampati, M.; Yadavilli, S.; Gordish-Dressman, H.; Santi, M.; Cruz, C.R.; Packer, R.J.; Almira-Suarez, M.I.; Hwang, E.I.; Nazarian, J. Differential Expression of Wilms’ Tumor Protein in Diffuse Intrinsic Pontine Glioma. J. Neuropathol. Exp. Neurol. 2019, 78, 380–388.
- Caretti, V.; Hiddingh, L.; Lagerweij, T.; Schellen, P.; Koken, P.W.; Hulleman, E.; Van Vuurden, D.G.; Vandertop, W.P.; Kaspers, G.J.L.; Noske, D.P.; et al. WEE1 Kinase Inhibition Enhances the Radiation Response of Diffuse Intrinsic Pontine Gliomas. Mol. Cancer Ther. 2013, 12, 141–150.
- Zanazzi, G.; Liechty, B.L.; Pendrick, D.; Krasnozhen-Ratush, O.; Snuderl, M.; Allen, J.C.; Garvin, J.H.; Mansukhani, M.M.; Roth, K.A.; Hsiao, S.J. Diffuse midline glioma with novel, potentially targetable, FGFR2–VPS35 fusion. Mol. Case Stud. 2020, 6.
- Tomita, T.; McLone, D.G.; Naidich, T.P. Brain stem gliomas in childhood. J. Neuro-Oncol. 1984, 2, 117–122.
- Xi, G.; Rajaram, V.; Mania-Farnell, B.; Mayanil, C.S.; Soares, M.B.; Tomita, T.; Goldman, S. Efficacy of vincristine administered via convection-enhanced delivery in a rodent brainstem tumor model documented by bioluminescence imaging. Child’s Nerv. Syst. 2012, 28, 565–574.
- Roujeau, T.; Machado, G.; Garnett, M.R.; Miquel, C.; Puget, S.; Geoerger, B.; Grill, J.; Boddaert, N.; Di Rocco, F.; Zerah, M.; et al. Stereotactic biopsy of diffuse pontine lesions in children. J. Neurosurg. Pediatr. 2007, 107, 1–4.
- Hashizume, R. Patient-derived Tumor Models for Diffuse Intrinsic Pontine Gliomas. Curr. Neuropharmacol. 2016, 15, 98–103.
- Caretti, V.; Zondervan, I.; Meijer, D.H.; Idema, S.; Vos, W.; Hamans, B.; Bugiani, M.; Hulleman, E.; Wesseling, P.; Vandertop, W.P.; et al. Monitoring of Tumor Growth and Post-Irradiation Recurrence in a Diffuse Intrinsic Pontine Glioma Mouse Model. Brain Pathol. 2010, 21, 441–451.
- Misuraca, K.L.; Cordero, F.J.; Becher, O.J. Pre-Clinical Models of Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2015, 5, 172.
- Lieberman, N.A.P.; DeGolier, K.; Kovar, H.M.; Davis, A.; Hoglund, V.; Stevens, J.; Winter, C.; Deutsch, G.; Furlan, S.N.; Vitanza, N.A.; et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: Implications for development of immunotherapy. Neuro-oncology 2018, 21, 83–94.
- Harris, W.; Newcomb, W.D. A Case of Pontine Glioma, with Special Reference to the Paths of Gustatory Sensation. Proc. R. Soc. Med. 1926, 19, 1–5.
- Pincus, D.W.; Richter, E.O.; Yachnis, A.T.; Bennett, J.; Bhatti, M.T.; Smith, A. Brainstem stereotactic biopsy sampling in children. J. Neurosurg. Pediatr. 2006, 104, 108–114.
- Broniscer, A.; Baker, J.N.; Baker, S.J.; Chi, S.N.; Geyer, J.R.; Morris, E.B.; Gajjar, A. Prospective collection of tissue samples at autopsy in children with diffuse intrinsic pontine glioma. Cancer 2010, 116, 4632–4637.
- Zarghooni, M.; Bartels, U.; Lee, E.; Buczkowicz, P.; Morrison, A.; Huang, A.; Bouffet, E.; Hawkins, C. Whole-Genome Profiling of Pediatric Diffuse Intrinsic Pontine Gliomas Highlights Platelet-Derived Growth Factor Receptor α and Poly (ADP-ribose) Polymerase as Potential Therapeutic Targets. J. Clin. Oncol. 2010, 28, 1337–1344.
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437.
- Nikbakht, H.; Panditharatna, E.; Mikael, L.G.; Li, R.; Gayden, T.; Osmond, M.; Ho, C.-Y.; Kambhampati, M.; Hwang, E.I.; Faury, D.; et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat. Commun. 2016, 7, 11185.
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 Activity by a Gain-of-Function H3 Mutation Found in Pediatric Glioblastoma. Science 2013, 340, 857–861.
- Mondal, G.; Lee, J.C.; Ravindranathan, A.; Villanueva-Meyer, J.E.; Tran, Q.T.; Allen, S.J.; Barreto, J.; Gupta, R.; Doo, P.; Van Ziffle, J.; et al. Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathol. 2020, 139, 1071–1088.
- Cohen, K.J.; Jabado, N.; Grill, J. Diffuse intrinsic pontine gliomas—Current management and new biologic insights. Is there a glimmer of hope? Neuro-Oncology 2017, 19, 1025–1034.
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pagès, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L.; et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827.
- Harutyunyan, A.S.; Krug, B.; Chen, H.; Papillon-Cavanagh, S.; Zeinieh, M.; De Jay, N.; Deshmukh, S.; Chen, C.C.L.; Belle, J.; Mikael, L.G.; et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 2019, 10, 1262.
- Chan, K.-M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990.
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.W.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA Hypomethylation Are Major Drivers of Gene Expression in K27M Mutant Pediatric High-Grade Gliomas. Cancer Cell 2013, 24, 660–672.
- Bechet, D.; Gielen, G.G.H.; Korshunov, A.; Pfister, S.M.; Rousso, C.; Faury, D.; Fiset, P.-O.; Benlimane, N.; Lewis, P.W.; Lu, C.; et al. Specific detection of methionine 27 mutation in histone 3 variants (H3K27M) in fixed tissue from high-grade astrocytomas. Acta Neuropathol. 2014, 128, 733–741.
- Piunti, A.; Hashizume, R.; Morgan, M.A.; Bartom, E.T.; Horbinski, C.M.; Marshall, S.A.; Rendleman, E.J.; Ma, Q.; Takahashi, Y.-H.; Woodfin, A.R.; et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017, 23, 493–500.
- Justin, N.; Zhang, Y.; Tarricone, C.; Martin, S.R.; Chen, S.; Underwood, E.; De Marco, V.; Haire, L.F.; Walker, P.A.; Reinberg, D.; et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 2016, 7, 11316.
- Castel, D.; Philippe, C.; Kergrohen, T.; Sill, M.; Merlevede, J.; Barret, E.; Puget, S.; Sainte-Rose, C.; Kramm, C.M.; Jones, C.; et al. Transcriptomic and epigenetic profiling of ‘diffuse midline gliomas, H3 K27M-mutant’ discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location. Acta Neuropathol. Commun. 2018, 6, 1–13.
- Nagaraja, S.; Vitanza, N.A.; Woo, P.J.; Taylor, K.R.; Liu, F.; Zhang, L.; Li, M.; Meng, W.; Ponnuswami, A.; Sun, W.; et al. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 31, 635–652.e6.
- Nagaraja, S.; Quezada, M.A.; Gillespie, S.M.; Arzt, M.; Lennon, J.J.; Woo, P.J.; Hovestadt, V.; Kambhampati, M.; Filbin, M.G.; Suva, M.L.; et al. Histone Variant and Cell Context Determine H3K27M Reprogramming of the Enhancer Landscape and Oncogenic State. Mol. Cell 2019, 76, 965–980.e12.
- Bailey, C.P.; Figueroa, M.; Gangadharan, A.; Yang, Y.; Romero, M.M.; A Kennis, B.; Yadavilli, S.; Henry, V.; Collier, T.; Monje, M.; et al. Pharmacologic inhibition of lysine-specific demethylase 1 as a therapeutic and immune-sensitization strategy in pediatric high-grade glioma. Neuro-Oncology 2020, 22, 1302–1314.
- Larson, J.D.; Kasper, L.H.; Paugh, B.S.; Jin, H.; Wu, G.; Kwon, C.-H.; Fan, Y.; Shaw, T.I.; Silveira, A.B.; Qu, C.; et al. Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell 2019, 35, 140–155.e7.
- An, S.; Camarillo, J.M.; Huang, T.Y.-T.; Li, D.; Morris, J.A.; Zoltek, M.A.; Qi, J.; Behbahani, M.; Kambhampati, M.; Kelleher, N.L.; et al. Histone tail analysis reveals H3K36me2 and H4K16ac as epigenetic signatures of diffuse intrinsic pontine glioma. J. Exp. Clin. Cancer Res. 2020, 39, 261.
- Pedersen, H.; Schmiegelow, K.; Hamerlik, P. Radio-Resistance and DNA Repair in Pediatric Diffuse Midline Gliomas. Cancers 2020, 12, 2813.
- Miyahara, H.; Yadavilli, S.; Natsumeda, M.; Rubens, J.A.; Rodgers, L.; Kambhampati, M.; Taylor, I.C.; Kaur, H.; Asnaghi, L.; Eberhart, C.G.; et al. The dual mTOR kinase inhibitor TAK228 inhibits tumorigenicity and enhances radiosensitization in diffuse intrinsic pontine glioma. Cancer Lett. 2017, 400, 110–116.
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450.
- Grabovska, Y.; Mackay, A.; O’Hare, P.; Crosier, S.; Finetti, M.; Schwalbe, E.C.; Pickles, J.C.; Fairchild, A.R.; Avery, A.; Cockle, J.; et al. Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat. Commun. 2020, 11, 4324.
- Lenzen, A.; Lauing, K.L.; Zhai, L.; Ladomersky, E.; Raman, P.; Rathi, K.; Lulla, R.R.; Hashizume, R.; A Wainwright, D. Novel RNA-targeting strategy for treating T cell-driven immunosuppression in human diffuse intrinsic pontine glioma. Neuro-oncology 2019, 21, ii92–ii93.
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat. Med. 2018, 24, 572–579.
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, eaaf2968.
- Shultz, L.D.; Ishikawa, F.; Greiner, D.L. Humanized mice in translational biomedical research. Nat. Rev. Immunol. 2007, 7, 118–130.
- Ito, R.; Takahashi, T.; Katano, I.; Ito, M. Current advances in humanized mouse models. Cell. Mol. Immunol. 2012, 9, 208–214.
- Sengupta, S.; Sobo, M.; Lee, K.; Kumar, S.S.; White, A.R.; Mender, I.; Fuller, C.; Chow, L.M.; Fouladi, M.; Shay, J.W.; et al. Induced Telomere Damage to Treat Telomerase Expressing Therapy-Resistant Pediatric Brain Tumors. Mol. Cancer Ther. 2018, 17, 1504–1514.
- Baiocchi, R.A.; Khatri, V.P.; Lindemann, M.J.; Ross, M.E.; Papoff, G.; Caprio, A.J.; Caprio, T.V.; Fenstermaker, R.; Ruberti, G.; Bernstein, Z.P.; et al. Phenotypic and Functional Analysis of Fas (CD95) Expression in Primary Central Nervous System Lymphoma of Patients with Acquired Immunodeficiency Syndrome. Blood 1997, 90, 1737–1746.
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.-E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 187–215.
- Biancotti, J.-C.; Town, T. Increasing Hematopoietic Stem Cell Yield to Develop Mice with Human Immune Systems. BioMed Res. Int. 2013, 2013, 740892.
- Meraz, I.M.; Majidi, M.; Meng, F.; Shao, R.; Ha, M.J.; Neri, S.; Fang, B.; Lin, S.H.; Tinkey, P.T.; Shpall, E.J.; et al. An Improved Patient-Derived Xenograft Humanized Mouse Model for Evaluation of Lung Cancer Immune Responses. Cancer Immunol. Res. 2019, 7, 1267–1279.
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014, 32, 364–372.

