 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Simone Patergnani | + 5895 word(s) | 5895 | 2021-04-12 06:09:26 | | | |
| 2 | Conner Chen | Meta information modification | 5895 | 2021-04-16 03:55:25 | | | | |
| 3 | Conner Chen | Meta information modification | 5895 | 2021-04-16 03:55:40 | | |
Video Upload Options
The word autophagy was introduced in late 1963 by the biochemist Christian de Duve [18] and defines a self-degradative cellular pathway whose intent is to degrade and recycle cellular contents. Autophagy exists in three forms that are classified according to their mechanisms and cellular functions: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). During microautophagy, the cytosolic material is wrapped and transported directly into the lumen of lysosomes. The main function of microautophagy (mA) is to control cell survival and organellar turnover upon nitrogen restriction. CMA has an important role in protein quality control (QC) and is responsible for degrading a specific subset of oxidized and damaged proteins. The selectivity of CMA is conferred by the existence of a specific pentapeptide motif (KFERQ), which is present in the amino acid sequences of all CMA substrates. Undoubtedly, the best-characterized and most prevalent form of autophagy in mammalian cells is macroautophagy (hereafter referred to as autophagy). Autophagy is responsible to capture a wide group of intracellular components, ranging from low-dimensional biological macromolecules to whole organelles, and bring them to the lysosomal compartment. Its physiological value rests on two main activities. On the one hand, autophagy acts as a QC mechanism that reshapes the cell, ensuring the removal of damaged proteins and organelles [27]. Selective forms of autophagy can specifically target mitochondria (mitophagy), the endoplasmic reticulum (ER; reticulophagy), peroxisomes (pexophagy), and lipid droplets (lipophagy).
1. A General Overview of Autophagy
The word autophagy was introduced in late 1963 by the biochemist Christian de Duve [1] and defines a self-degradative cellular pathway whose intent is to degrade and recycle cellular contents. Autophagy exists in three forms that are classified according to their mechanisms and cellular functions: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). During microautophagy, the cytosolic material is wrapped and transported directly into the lumen of lysosomes. The main function of microautophagy (mA) is to control cell survival and organellar turnover upon nitrogen restriction. Unfortunately, due to the lack of specific methods for measuring mA (apart from electron microscopy), the effective contributions of mA in mammalian cells remain little studied, and most studies about mA molecular processes are carried out in yeast [2]. Despite this, different investigations suggest that the molecular dynamics of mA existing in yeast may be conserved in mammalian mA. Consistently with this, it has been demonstrated that the endosomal sorting complex required for transport (ESCRT) system is involved in mammalian [3] and yeast mA [4]. Furthermore, a prolonged starvation condition [5][6] as well as cellular treatments with the macrolide compound rapamycin activates mA in both mammalian and yeast cells [4][7][8].
CMA has an important role in protein quality control (QC) and is responsible for degrading a specific subset of oxidized and damaged proteins. The selectivity of CMA is conferred by the existence of a specific pentapeptide motif (KFERQ), which is present in the amino acid sequences of all CMA substrates. This motif is identified by the cytosolic chaperone heat shock-cognate protein of 70 kDa (hSC70), which brings the protein target directly to the lysosome surface [9]. In the last decade, several advances have been made in understanding the molecular mechanisms of CMA. These findings suggest an important contribution of CMA to diverse human diseases, including neurodegeneration [9]. Undoubtedly, the best-characterized and most prevalent form of autophagy in mammalian cells is macroautophagy (hereafter referred to as autophagy), whose multistep process and contribution to the pathophysiology of diverse neurodegenerative conditions will be discussed throughout this review.
Autophagy, a complex intracellular process that is very ancient and has been strongly conserved during evolution, exists to identify and capture a wide group of intracellular components, ranging from low-dimensional biological macromolecules to whole organelles, and bring them to the lysosomal compartment. Its physiological value rests on two main activities. On the one hand, autophagy acts as a QC mechanism that reshapes the cell, ensuring the removal of damaged proteins and organelles [10]. Selective forms of autophagy can specifically target mitochondria (mitophagy), the endoplasmic reticulum (ER; reticulophagy), peroxisomes (pexophagy), and lipid droplets (lipophagy). In addition, autophagy participates in the struggle against invading pathogens (xenophagy), inducing cell defense [11].
On the other hand, lysosomal degradation represents an important source of amino acids and lipids for the de novo synthesis of proteins and lipids. This is of particular importance during starvation, which limits amino acid availability. The limited availability of amino acids affects protein synthesis, which can be performed only in the presence of all the necessary building blocks, in particular, essential amino acids. Under shortage conditions, amino acid pool completeness can be fulfilled only through the degradation of cellular proteins. In such a way, autophagy represents a fundamental survival mechanism, particularly during stress conditions originating from hypoxia or pathogen invasion [10].
Thus, it is not surprising that energy availability can regulate or trigger autophagy and, in particular, that a large number of stimuli converge on metabolic energy sensors, such as mammalian target of rapamycin (mTOR) and 5′ adenosine monophosphate-activated protein kinase (AMPK), which, in turn, regulate autophagy [12].
In cells, mTOR exists in two complexes, mTORC1 and mTORC2, which not only are composed of different protein-binding partners but also regulate different pathways. The primary role of mTORC2 is to regulate cell survival and cytoskeletal organization, while its role in autophagy remains poorly understood. Recent work has shed light on this obscure point. Indeed, the transforming growth factor beta (TGFB)/INHB/activin signaling pathway has been recently identified as an upstream regulator of mTORC2. TGFB-INHB/activin mediates mTORC2 inhibition and regulates the autophagic flux and the cardiac functions in a Drosophila cardiac-specific knockdown of TGFB-INHB/activin model [13]. Another investigation recently confirmed the importance of mTORC2 for autophagy. In this case, it has been demonstrated that mTORC2 exists on a molecular axis with the serum- and glucocorticoid-inducible kinase 1 (SGK-1) and, in this state, controls autophagy and mitophagy induction. Consistently, mTORC2- or SGK-1 deficient C. elegans models present a perturbed mitochondrial homeostasis and aberrant ROS production, which trigger autophagy and mitophagy. Excessive autophagic and mitophagic fluxes, in turn, result in developmental and reproductive deficits in mTORC2- or SGK-1-deficient animals [14]. Oppositely, the primary role of mTORC1 is to play a pivotal role in cellular catabolic pathways, particularly autophagy [15]. To exert its function, mTORC1 integrates different stimuli, including hormonal stimulation, nutrient availability, and the oxygen level. In the presence of normal levels of energy and amino acids, mTOR inhibits autophagy through specific unc-51-like autophagy-activating kinase 1 (ULK1) serine phosphorylation at the phosphorylation site Ser 757. By contrast, in response to nutritional deprivation, oxygen unavailability, and mitochondrial dysfunction, AMPK activates autophagy through the phosphorylation of ULK1 at Ser 317 and Ser 777 [16]. Interestingly, another research group demonstrated that AMPK may phosphorylate ULK in additional sites. Indeed, by employing a bioinformatic approach, it has been found that ULK1 contains a further four potential AMPK sites [17]. Three of them (Ser 555, Ser 637, and Thr 574) were also identified by mass spectrometry in cells pretreated with an AMPK activator, while the site Ser 467 was confirmed by immunoblotting with phosphospecific antibodies [17]. Unfortunately, this work lacks an analysis of the effect of the different phosphorylations on autophagy. By using SILAC (stable isotope labeling with amino acids) technology, other work mapped 13 new phosphorylation sites of ULK1 [18]. All of them were dependent on nutrient availability, but only Ser 638 and Ser 758 displayed the most significant changes. In addition, time course experiments investigating the response to nutrient availability demonstrated that these phosphorylations were differentially regulated and that mTOR mediated both phosphorylations. Intriguingly, the authors also demonstrated that the phosphorylation at Ser 638 was also mediated by AMPK [18]. Altogether, these findings demonstrate that ULK1 is the key regulator of autophagy, and the occurrence of different protein phosphorylation events is crucial for regulating its activity. Furthermore, the concurrent existence of at least two opposite regulatory pathways that converge on ULK1 signaling (mediated by MTOR and AMPK) allows the cell to better adapt to extracellular and intracellular variations but also affects several pathological conditions.
In the cells, ULK1 forms a complex with autophagy-related (ATG) 13/200-kDa focal adhesion kinase family-interacting protein (FIP200) and ATG101. As reported above, ULK1 activity is mainly regulated by phosphorylation/desphosphorylation events mediated by AMPK and mTOR. In addition, it has been demonstrated that ULK1 is able to phosphorylate itself at Thr 180 [19] and FIP200, ATG13, and ATG101 [20][21] and that the phosphorylation events are regulated by protein phosphatase. Protein phosphatase 2A (PP2A) and protein phosphatase 1D magnesium-dependent delta isoform (PPM1D) regulate the ULK1 phosphorylation [22][23]. PP2C phosphatases (Ptc2 and Ptc3) mediate the dephosphorylation of ATG13 30655342. The ULK1/ATG13/FIP200/ATG101 molecular axis represents the most upstream regulatory complex related to double-membrane vacuole (autophagosome) formation [12]. Autophagosomes symbolize the starting moment of the whole autophagic process, which begins with the formation of double-membrane lined vesicles that fuse together to engulf portions of the cytoplasm. The resulting double-membrane vacuoles are autophagosomes, which can fuse with vesicles of the endocytic pathway at different stages of maturation or directly with lysosomes, becoming autolysosomes. In autolysosomes, acidic hydrolases break down macromolecules into smaller constituents that are released back to the cytosol by lysosomal transporters and permeases. Once activated, the ULK1/ATG13/FIP200/ATG101 molecular axis also phosphorylates and activates coiled-coil, moesin-like BCL2 interacting protein (BECN1) [24][25]. BECN1 can be part of a complex including class III phosphatidylinositol 3-kinase (PI3K) and its regulatory proteins vacuolar protein sorting 34 (Vsp34), p150, and ATG14L. Upon activation, this complex is involved in the nucleation and elongation of autophagosomes. The first step occurs on the surface of the membranes of the ER, mitochondria, Golgi complex, endosomes, or plasma membrane [26] and consists of the phosphorylation of phosphatidylinositol to form phosphatidylinositol-3-phosphate (PI3P). This phosphoinositide behaves as a positive regulator of autophagy. In fact, the presence of PI3P at the source membrane triggers the docking of several adaptor proteins, which, in turn, induce and sustain the elongation of the sack-like, omega-shaped structure, which grows, binds, and surrounds the material intended to be digested.
Another interaction of BECN1 can exert an inhibitory effect on autophagy [27]. BECN1 has been reported to bind B-cell lymphoma (BCL)-2, BCL-XL, and other members of the BCL-2 family through the BCL-2-homology-3 (BH3) domain. The consequence of this interaction is a diminution of the interaction between BECN1 and the class III PI3K complex, which prevents the formation of phagophores [27]. Accordingly, BCL-2 phosphorylation can reverse BECN1 sequestration and restore autophagy stimulation [27].
The other two systems, ATG12–ATG5–ATG16L1 and microtubule-associated protein 1A/1B-light chain 3 (LC3)–phosphatidylethanolamine (PE) complexes, seem to play an important role in the elongation and closure of autophagosomes, although the underlying mechanism has not yet been clarified. A key process during autophagosome elongation and closure is the lipidation of the LC3 protein, which is joined to the membrane PE. Once inserted into the autophagosomal membrane, the lipidated complex can further recruit other adaptor proteins. This allows autophagosomes to recognize cargo material, and elongate and close the vesicle. The fusion of the autophagosomes with the lysosome is the subsequent step, which, in a normally operating lysosome, is followed by lysosomal compartment acidification, the degradation of macromolecules by hydrolases and lipases, and the recycling of the base constituents (Figure 1).
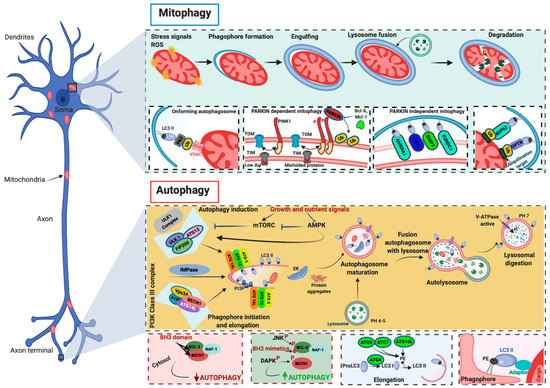
Figure 1. Molecular mechanisms of autophagy and mitophagy. The mammalian target of rapamycin (mTOR) and the 5′ adenosine monophosphate-activated protein kinase (AMPK) are the main negative and positive regulators of autophagy, respectively. One of the primary targets of the action of mTOR and AMPK is the unc-51-like autophagy-activating kinase 1 (ULK1)/autophagy-related (ATG) 13/FIP200 (200-kDa focal adhesion kinase family-interacting protein) complex, which is the main regulator of autophagosomal formation. Other important proteins that participate in this molecular process are the coiled-coil, moesin-like BCL-2 interacting protein (BECN1), class III phosphatidylinositol 3-kinase (PI3K), vacuolar protein sorting 34 (Vsp34), ATG14L, p150, and IMPase. The activity of BECN1 in regulating the autophagy process is also mediated by the interaction with BCL-2. During the elongation of the autophagosome, a series of autophagy-related (ATG) proteins are involved. In particular, two specific complexes were found to be essential for completing autophagosomal formation: (ATG)12–ATG5–ATG16L1 and microtubule-associated protein 1A/1B-light chain 3 (LC3)–phosphatidylethanolamine (PE) complexes. Mitochondria are particularly vulnerable to stress signals, such as ROS, which, in turn, can cause severe mitochondrial dysfunction and activate the mitophagic process. PINK1 senses this mitochondrial damage and phosphorylates and recruits Parkin to the outer mitochondrial membrane of the mitochondria. Phosphorylation converts Parkin to an active ubiquitin (Ub)-dependent enzyme and mediates the phosphorylation of different mitochondrial proteins. During this process intervene different Ub-binding autophagy receptors such as p6, NBR1, NDP52, and optineurin (OPTN), which connect the damaged mitochondria to the forming autophagosomes. Mitophagy may also be executed in a Parkin-independent manner. In this case, different proteins (FUNDC1, AMBRA1, NIX, and BNIP3) intervene to signal the mitochondria that should be degraded.
2. Mitophagy: The Master Regulator of the Mitochondrial Population
Mitochondria are essential intracellular organelles that supply substrates and energy to execute numerous cell functions, such as metabolism, differentiation, apoptosis, cell movement, and differentiation. In contrast to other intracellular components, mitochondria are constituted by two membranes, the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM), which fully surround the mitochondrial matrix. Between the OMM and IMM, another mitochondrial subcompartment exists, the intermembrane space (IMS) [28]. Another unique feature of mitochondria is that they have their own genome (mitochondrial DNA, mtDNA), which encodes 13 proteins that are essential components of the oxidative phosphorylation (OXPHOS) system, the process by which ATP is formed [29]. A series of members (complexes I-V, C-I-V) of the mitochondrial electron chain (mETC) found in the IMM permit the transfer of electrons from NADH or FADH2 to O2 [30]. The energy produced during this movement creates a proton gradient that is used by the last component of the mETC (C-V, ATP synthase) to synthesize ATP [30]. The impairment of electron transfer or stress conditions affect the production of reactive oxygen species (ROS), of which C-I and C-III are the main producers [31]. Mitochondria are also central hubs for calcium (Ca2+) signaling [32]. At rest, mitochondria have low Ca2+ concentrations [Ca2+] (~100 nM range or lower). However, upon stimulation, mitochondrial [Ca2+] can increase to the range of hundreds in micromolar concentration [33]. This happens due to the highly specialized contact sites (mitochondria-associated membranes, MAMs) that exist between mitochondria and the main intracellular Ca2+ store of cells, the ER [34]. These interaction sites represent critical hubs for the regulation of diverse cellular processes (such as energy metabolism, inflammation, redox regulation, and lipid and protein transfer), and recently, MAMs have been described to play an important role in the onset and progression of several human diseases by regulating Ca2+ transmission between the ER and mitochondria [35]. Once released from the ER, Ca2+ can enter mitochondria owing to the close proximity of the ER to mitochondria, the electrochemical driving force (mitochondrial membrane potential) that is created by electron transfer, and the activity of the components of the mitochondrial Ca2+ uniporter (MCU) [36][37]. Mitochondria are normally present in cells in the form of a dynamic network, where the mitochondrial mass increases as a consequence of mitochondrial biogenesis. The control and reshaping of the mitochondrial population can occur through different mechanisms [38]. These mechanisms include (i) the control of protein quality through mitochondrial proteases, the mitochondrial unfolded protein response, or proteasome-dependent degradation; (ii) the budding of mitochondrion-derived vesicles; and (iii) the targeting of some or all mitochondria to lysosomes through mitophagy.
Mitophagy regulation is not yet a completely understood process. During short-term starvation, the mitochondrial pool is not depleted, so as to not further reduce the cellular production of energy, while oxidative metabolism is mainly sustained by general autophagy [12]. This fact necessarily implies a difference in regulation between autophagy and mitophagy that allows the cautious sparing of mitochondria, which are among the principal end-users of the material provided by autophagy. A role in this sense seems to be played by fission restriction. In fact, fragmented mitochondria appear to be a preferred target for mitophagy: when their number is reduced, mitophagy itself is restricted.
When the ultimate goal is to eliminate mitochondria, there are different physiological mechanisms that can be activated. The first example is programmed mitophagy. There are several situations in the cell that can require the activation of programmed mitophagy, independent of the wellness of mitochondria. An example is the mitochondrial depletion that occurs in reticulocytes during differentiation through the activity of NIP3-like protein X (NIX/BNIP3L). Other examples include the elimination of male-derived mitochondria after egg fertilization [39] and the reshaping of the mitochondrial population during cardiomyocyte [40] or muscle cell differentiation, which induces a change from carbohydrate- to fatty acid-driven OXPHOS [41]. Stimulations that can normally trigger mitophagy can be affected by mitochondrial defects, such as a decline in transmembrane potential and excessive ROS production.
Mitophagy involves some fundamental steps. First, as stated above, mitochondria must assume the dimensions necessary to easily enter autophagosome vesicles. Therefore, they are normally resized through fission processes. In addition, they need to be properly displayed on the surface to trigger the formation of vesicles, which will engulf them. Typically, “eat-me signals” can be ubiquitin-dependent or not. The best-known example of a ubiquitin-dependent mechanism is the PTEN-induced kinase 1 (PINK1)/Parkin axis. PINK1 and Parkin belong to a series of genes referred to as PARK genes, which include α-syn (PARK1/4), Parkin (PARK2), PINK1 (PARK6), protein deglycase-1 (DJ-1, PARK7), leucine-rich repeat kinase 2 (LRRK2, PARK8), and ATP13A2 (PARK9). The name of this group of genes (Parkin genes) comes from the finding that mutations in these genes have been linked to familiar forms of PD. In particular, approximately 100 mutations in the Parkin gene have been identified as causing autosomal recessive Parkinsonism [42].
PINK1 is a mitochondrial serine/threonine-protein kinase, and Parkin is an E3 ubiquitin ligase; these proteins induce different functions at the cellular level but act in a common pathway to regulate mitophagy.
PINK1 is a ubiquitous protein characterized by a mitochondrial targeting sequence (MTS), a transmembrane domain, and a highly conserved serine/threonine kinase domain. At present, approximately 30 pathogenic PINK1 mutations that impair its kinase activity and provoke loss of function have been identified [43][44][45][46].
Normally, PINK1 is imported into mitochondria via the activity of the translocase of the inner membrane (TIM)–translocase of the outer membrane (TOM) complex. Once PINK1 arrives in the IMM, it is subjected to a series of proteolytic cleavages that reduce the full-length form of PINK1 into fragments, which are then degraded by the proteasome [47][48][49]. In the presence of alterations in mitochondrial membrane potential, the activity of the TIM/TOM complex is reduced, and PINK1 begins to accumulate on the OMM. Here, after being stabilized by a molecular complex including TOM proteins [50][51], PINK1 phosphorylates Parkin. The phosphorylation converts Parkin from an autoinhibited enzyme to an active ubiquitin (Ub)-dependent enzyme [52][53]. In this state, Parkin actively ubiquitinates several mitochondrial proteins at the OMM. The ubiquitination events promote the recruitment of the Ub-binding autophagy receptors p62/Sequestome, NBR1, NDP52, optineurin (OPTN), and TAX1BP1 (TBK1), which connect damaged mitochondria to phagosomes for clearance in lysosomes [54][55][56]. In recent years, different studies have identified pathways regulating mitophagy that are PINK1–Parkin-independent. These mechanisms may act in parallel or in addition to PINK1–Parkin-dependent mitophagy and involve a series of OMM mitophagy receptors that bind LC3 and recruit mitochondria to autophagic vesicles. Among them, the most studied are the proapoptotic members of the BCL2 family, NIX and BNIP3 [57][58] and FUNDC1 [59], which regulate the mitophagy process during ischemic/hypoxic conditions, and the BECN1 regulator AMBRA1. Interestingly, it has been proven that AMBRA1 regulates both Parkin-dependent and Parkin-independent mitophagy [60] (Figure 2).
3. Relationship between Autophagy and Mitophagy in MS
Multiple sclerosis (MS) is a progressive and chronic disease that affects approximately 3 million persons worldwide. MS is an inflammatory condition in which activated immune cells enter the central nervous system (CNS) and cause progressive demyelination, gliosis, and neuronal loss. The symptoms vary from individual to individual [61]. The most common symptoms are walking difficulties, sensory disturbances, vision problems, and cognitive and emotional impairments. Typically, MS starts with an unexpected onset of neurological impairments, and the majority of individuals display a relapsing–remitting (RR) course of the disease in which recurrent periods alternate with relapse phases. This course may be followed by a secondary progressive phase in which inflammatory attacks are more frequent and cause irreversible neurological impairments. A small percentage of individuals may present with the primary progressive form of the disease, which is characterized by the absence of remission periods and a progressive worsening of symptoms [62]. Currently, the pathogenesis and etiology of MS are unclear. MS is considered a multifactorial disease, and genetic predisposition and environmental factors may play important roles in disease progression. Furthermore, mitochondrial dysfunction as well as the impairment of the QC systems of mitochondria have been identified in different MS samples and represent evidence that the mitochondrial compartment has a major role in MS [63]. In addition, recent investigations have described an important contribution of autophagic processes. The first evidence that autophagy could be involved in MS was reported in 2009, when a strong correlation was found between the expression of the autophagic marker ATG5 and the clinical disability observed in the experimental autoimmune encephalomyelitis (EAE) MS animal model. Moreover, in this work, the authors found increased expression of ATG5 in T cells obtained from RR-MS patients and in postmortem brain tissue from individuals with secondary progressive MS [64]. Unfortunately, the authors did not address the role of autophagy in T cells and MS. They only speculated that autophagy may help to increase the survival of T cells and help to propagate the immune response. Similarly, other work detected ATG5 increases in terms of both mRNA levels and protein amounts in T cells obtained from MS patients who were treatment naïve [65]. Increases in ATG5 also correlated with the presence of proinflammatory cytokines, thus displaying a possible relationship between the inflammatory status and ATG5 expression in MS. However, they did not perform a detailed analysis of the clinical activity state [65]. T cells present different subpopulations. Among them, T regulatory cells (Treg) are particularly relevant in autoimmune disease because they prevent inflammation and preserve the tolerance to self-antigens. Recently, it has been demonstrated that the autophagic mediator AMBRA1 associates with the protein phosphatase PP2A to sustain Treg differentiation by increasing the expression of Forkhead box P 3 (FOXP3), an essential transcription factor for the differentiation of Treg cells [66]. In addition, the AMBRA1–PP2A–FOXP3 molecular axis was found to be essential for regulating the optimal autophagic levels necessary for T-cell stimulation and differentiation. Consistently, AMBRA1 conditional KO mice display reductions in FOXP3 levels with consequent impairments in Treg differentiation and activity. Most importantly, AMBRA1 deficiency worsens the disease pathogenesis in an EAE MS animal model [66]. Finally, work of Akatsuka et al. not only demonstrates the important role of AMBRA1 in the regulation of T cells, but also highlights decreased mitochondrial functioning and metabolism in these cells [67]. All these findings demonstrate that AMBRA1 is an essential factor that regulates both autophagic and mitochondrial behaviors and, probably, also the mitophagic process in T cells.
In MS, T-cell activities may be modulated by the complement-regulating molecule CD46 [68]. This factor is also described as an autophagic inducer [69], and its levels are documented to be increased in the serum and cerebrospinal fluid (CSF) of MS patients [70]. The increased T-cell autoreactivity in MS may also be promoted by IRGM1, a GTPase that regulates the survival of immune cells through autophagy. Consistent with this finding, IRGM1 deletion increases the apoptosis of T cells, reduces their proliferative capacity, and ameliorates the clinical score of the EAE mouse model [71]. Considering that subsequent studies have demonstrated that IRGM1 is localized to the mitochondrial compartment and regulates the mitochondrial metabolism and mitochondrial fission induced by mitophagy [72][73], the increased T-cell autoreactivity observed in MS may be due to an impairment in the mitophagic process. In addition to its effects on T cells, autophagy plays a role in dendritic cells (DCs), the most potent antigen-presenting cells (APCs) in the immune system. In particular, autophagy starts in response to bacterial and viral infection. By generating transgenic mice with silencing of ATG7 in DCs, Bhattacharya and colleagues demonstrated the importance of DCs and autophagy in MS. Indeed, they showed that the specific loss of autophagy in DCs significantly delayed disease progression and reduced disease severity in EAE mice [74]. As reported above, AMPK is the main positive regulator of autophagy. This kinase works by sensing the AMP/ATP ratio and activates autophagy to combat energetic imbalance. It has been demonstrated that following exposure to proinflammatory cytokines, AMPK activates and triggers autophagy in oligodendrocyte precursor cells (OPCs) [75]. This change is due to a metabolic switch from OXPHOS to glycolysis and impairment of mitochondrial dynamics, leading to increased oxidative stress and reduced mitochondrial Ca2+ uptake and ATP production. As a consequence, OPCs fail to differentiate into mature and myelinating oligodendrocytes [75]. In support of these in vitro findings, recent work demonstrated that metabolic stress-induced autophagy is a key element in an in vivo MS model. Indeed, MCU-deficient (MCU-def) mice subjected to EAE displayed elevated clinical scores, excessive inflammation, and demyelination [76]. Morphological and functional analyses performed with the spinal cords of MCU-def mice revealed important mitochondrial damage, accompanied by an elevated presence of autophagosomal markers and a decrease in ATP synthesis and mitochondrial gene expression. Overall, these data confirm that the presence of mitochondrial dysfunction provokes the inhibition of Ca2+ buffering, ATP synthesis, and mitochondrial gene expression, causing a metabolic collapse that prompts autophagy and worsens MS-like conditions. Furthermore, since autophagic activation accompanied by the downregulation of PGC1α (a master regulator of mitochondrial biogenesis) has been observed, it is possible to speculate that the mitochondrial QC system is also affected. However, studies have not verified whether autophagy activities lead to autophagic mitochondrial removal.
Markers of autophagic processes may represent reliable potential biomarkers for monitoring the progression of disease. Increased amounts of Parkin, ATG5, and inflammatory cytokines are present in both the serum and CSF obtained from MS patients. Analyses comparing MS patients to healthy individuals and patients affected by other neurodegenerative conditions have been conducted [77]. Moreover, subsequent work demonstrated that increases in both autophagic and mitophagic markers correlated with the active phases of the disease and with circulating lactate levels, demonstrating the presence of an impaired metabolic status in MS patients [78]. Notably, several studies have associated lactate levels with MS progression [79]. Other independent research groups have confirmed that circulating autophagy and mitophagy markers are increased in MS biofluids [80][81]. In addition, the circulating levels of mitochondrial adenine nucleotide translocase 1 (ANT1) and oxidative stress markers have also been investigated. Interestingly, MS patients display increased oxidative stress, accompanied by reduced levels of the mitochondrial marker ANT1, suggesting that the mitochondrial QC systems are activated to promote the removal of nonfunctioning mitochondria. Consistent with this, reduced circulating levels of the OMM protein translocator protein 18 kDa (TSPO) and increased amounts of the mitochondrial disease marker growth/differentiation factor 15 (GDF-15) have been found in MS individuals and correlate with the severity of the disease [82][83] (Table 1).
Table 1. Summary of autophagy- and mitophagy-related markers in biofluids of MS-, AD-, and PD-affected persons.
| Neurodegenerative Condition | Marker | Role | Type of Human Biofluid |
|---|---|---|---|
| MS | Parkin | Mitophagy regulator | Serum, CSF |
| ATG5 | Autophagy regulator | Serum, CSF | |
| Mitochondrial adenine nucleotide translocase 1 (ANT1) | Mitochondrial ADP/ATP translocase | Serum, CSF | |
| Translocator protein 18 kDa (TSPO) | Regulator of mPTP opening | Blood PBMCs | |
| Growth/differentiation factor 15 (GDF-15) | Mitochondrial disease marker | Serum | |
| TNFα | Proinflammatory cytokine | Serum, CSF | |
| Lactate | Mitochondrial dysfunction marker | Serum, CSF | |
| AD | BECN1 | Autophagy regulator | Blood PBMCs, serum |
| p62 | Autophagy regulator | Blood PBMCs | |
| LC3 | Autophagy regulator | Blood PBMCs | |
| ATG5 | Autophagy regulator | Plasma, serum | |
| Parkin | Mitophagy regulator | Serum | |
| EEA1, LAMP1, LAMP2, RAB3, and RAB7 | Lysosomal regulators | CSF | |
| PD | LC3B | Autophagy regulator | CSF |
| BECN1 | Autophagy regulator | CSF, blood PBMCs | |
| ATG5 | Autophagy regulator | CSF | |
| LAMP2 | Lysosomal regulator | CSF | |
| ULK1 | Autophagy regulator | Blood PBMCs | |
| ATG5 | Autophagy regulator | Blood PBMCs | |
| ATG4B | Autophagy regulator | Blood PBMCs | |
| ATG16L1 | Autophagy regulator | Blood PBMCs |
It is clear that autophagy and mitophagy as well as the mitochondrial quality control system are important contributors in MS. In the last few years, an increasing number of studies have correlated the activities of such molecular mechanisms with the progression of the disease. Furthermore, circulating elements of autophagy and mitophagy may be detected in human samples from MS individuals, thus suggesting the possibility of using them as novel biomarkers. However, MS shows a great heterogeneity with regard to the clinical symptoms as well as therapy response. In addition, MS manifests in different forms (clinically isolated syndrome, RR MS, secondary progressive MS, and primary progressive MS), where the relapse rate and disability progression differentiate one from the other. Only when the dynamics and response of autophagy and mitophagy are well characterized in regard to all these conditions will we be able to claim to have identified the real contributions of them in MS, and we could use autophagic and mitophagic elements as innovative markers for MS disease progression.
4. Involvement of Autophagy Mechanisms in AD Progression
AD was first described in the early 20th century and is characterized by a progressive deterioration of cognitive function. Memory loss and dementia represent the most common symptoms. The cardinal pathological hallmarks of AD are extracellular (amyloid) plaques and intracellular and extracellular neurofibrillary tangles (NFTs). Amyloid plaques are composed of deposits of Aβ, α-syn, Ub, and apolipoprotein E. NFTs are characterized by hyperphosphorylated tau protein and apolipoprotein E. These aggregates induce neuronal toxicity by impeding neural communication and provoking cell death either directly or by preventing the delivery of an optimal nutrient supply to brain cells [84].
At present, the origin of AD and the mechanisms occurring in the pathogenesis of AD are not well defined. Inflammation seems to play an important role: mediators of inflammation, such as cytokines, adhesion molecules, and prostaglandins, drive degeneration in different neural AD models [85]. Consistent with this finding, aggregated peptides increase proinflammatory agent production, and inflammatory molecules are detected in the CSF, serum, and plaques obtained from AD patients. Oxidative and nitrosylative damage provoked by ROS and reactive nitrogen species (RNS) are determinants of the initiation and progression of AD [86]. Oxidatively damaged membrane phospholipids and increased oxidative stress in neurons are frequently present in neurons exposed to Aβ [87]. Furthermore, AD brains extracted at autopsy have decreased amounts of vitamins A and E and β-carotene [88] and display a higher production of free radicals and increased expression of neuronal nitric oxide synthase (nNOS) [89]. This increased nNOS correlates with an increased apoptosis of hippocampal neurons. In the last 10 years, an increasing number of studies have demonstrated the critical contributions of autophagy and mitophagy to AD pathogenesis [90]. Several studies have reported an increased presence of Aβ in autophagosomes [91]. Interestingly, autophagosomes also contain amyloid precursor protein (APP) and its processing enzymes, in particular, a component of the γ-secretase complex, suggesting an additional source of Aβ. Consistent with this finding, the induction of autophagy correlates with Aβ production, and autophagy-deficient animals (with ATG7 knockdown) display reduced Aβ secretion [92]. Additionally, the hyperphosphorylation of tau correlates with increased autophagic levels. Indeed, postmortem AD brain samples are characterized by LC3- and p62-positive autophagosomes, and the hyperphosphorylation of tau has been recognized in autophagy-deficient mice [93][94]. Although these observations highlight a dangerous correlation between autophagy and AD, other studies suggest that autophagy and mitophagy may exert beneficial effects against AD [90]. The abnormal accumulation of autophagosome vesicles is present in AD neurons [91]. This accumulation is related to compromised lysosomal function, which results in lysosomes that are no longer able to degrade autophagosomes. The overexpression of Parkin and PINK activates mitophagy, restores mitochondrial function, and reduces Aβ production [95][96]. Similar results have been obtained from another independent experiments that demonstrated that mitophagy is essential for reducing Aβ levels, abolishing tau hyperphosphorylation, preventing cognitive impairments in an AD mouse model, and suppressing neuroinflammation [97].
To confirm the crucial role of autophagic and mitophagic dynamics in AD, different studies have evaluated the presence of elements belonging to these processes in biofluids from persons with AD. The first investigation was performed in 1995, in which ventricular CSF from postmortem AD patients was analyzed. In this study, the authors detected increased levels of the lysosomal protein cathepsin D [98]. However, a subsequent report performed with lumbar CSF samples from living AD patients found no change in the levels of cathepsin B [99]. This finding was confirmed in other work that investigated a broad range of lysosomal proteins in CSF samples from living AD patients and found no variations in diverse cathepsin forms (A, B, D, and L); however, the study did find altered expression for five other lysosomal proteins in the AD samples: early endosomal antigen 1 (EEA1), LAMP1, LAMP2, RAB3, and RAB7 [100]. By contrast, a recent study analyzed the levels of proteins associated with lysosomal function in the CSF of AD persons by conducting solid-phase extraction and parallel reaction monitoring mass spectrometry and found only minor or absent changes in their levels [101]. Unfortunately, the levels of proteins directly related to autophagy and mitophagy processes were not investigated in that study. A follow-up study at 12 and 24 months identified autophagic elements (BECN1, p62, and LC3) in peripheral blood mononuclear cells (PBMCs) obtained from the blood of AD patients and demonstrated that their levels varied during the course of the disease and correlated with the inflammatory environment [102]. Recently, autophagic elements have also been assessed directly in AD blood samples. Indeed, increased levels of the autophagic marker ATG5 are present in the plasma of patients with dementia who meet the criteria for probable AD. Unfortunately, the authors did not identify the subtype of dementia or confirm the AD status. These limitations were overcome in a recent investigation assessing the circulation of autophagic and mitophagic markers in the serum of patients affected by mild–moderate late-onset AD, mild cognitive impairment (MCI), vascular dementia (VAD), and mixed dementia (MD). In this work, the authors found decreased levels of ATG5 and Parkin in patients affected by AD, MCI, and MD. By contrast, they detected increased levels of these markers in VAD patients [103]. This investigation suggests that autophagy and mitophagy markers are possible biomarkers for AD and that they are differentially affected in different dementia types, which may help to discriminate AD-type dementias from VAD. Additionally, the fact that AD samples have decreased levels of autophagy and mitophagy markers confirms the presence of an impaired degradative system in AD persons (Table 1).
Summing up, autophagy and mitophagy represent well-established mechanisms in AD and may exert a protective role. Accordingly, most research highlights the reduced recruitment of both autophagy and mitophagic factors in cell cultures, in vivo AD models, and human samples obtained from AD-affected patients, including in the body fluids of the CSF and blood. Here, autophagic and mitophagic partners also correlate with the inflammatory status and change during the course of the disease, thus opening up the possibility of using autophagic and mitophagic elements as markers for the progression of AD. However, before ascribing merit to these molecules as potential screening, prognostic, diagnostic, or disease-monitoring markers for AD, it is important to consider different aspects. The diagnosis of AD cannot be achieved until the patient displays dementia symptoms. In addition, different dementia types exist and vary between individuals. Very few studies have monitored the variation of circulating markers of autophagy and mitophagy during the different dementia types. Furthermore, these studies lack validation of the investigated markers with accepted methods for diagnosing AD, such as amyloid PET imaging. Again, all the investigations performed did not provide follow-up studies and did not analyze the effects of the disease-modifying drugs commonly used for AD therapy on autophagy and mitophagy circulating markers. Undoubtedly, more detailed analyses and larger cohort studies are necessary to verify whether autophagic and mitophagic circulating elements may represent promising biomarkers for AD.
References
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492.
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682.
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139.
- Morshed, S.; Tasnin, M.N.; Ushimaru, T. ESCRT machinery plays a role in microautophagy in yeast. BMC Mol. Cell Biol. 2020, 21, 70.
- Sato, M.; Seki, T.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H. Fluorescent-based evaluation of chaperone-mediated autophagy and microautophagy activities in cultured cells. Genes Cells Devoted Mol. Cell. Mech. 2016, 21, 861–873.
- Olsvik, H.L.; Svenning, S.; Abudu, Y.P.; Brech, A.; Stenmark, H.; Johansen, T.; Mejlvang, J. Endosomal microautophagy is an integrated part of the autophagic response to amino acid starvation. Autophagy 2019, 15, 182–183.
- Sato, M.; Seki, T.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H. Rapamycin activates mammalian microautophagy. J. Pharmacol. Sci. 2019, 140, 201–204.
- Rahman, M.A.; Terasawa, M.; Mostofa, M.G.; Ushimaru, T. The TORC1-Nem1/Spo7-Pah1/lipin axis regulates microautophagy induction in budding yeast. Biochem. Biophys. Res. Commun. 2018, 504, 505–512.
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381.
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364.
- Patergnani, S.; Pinton, P. Mitophagy and mitochondrial balance. Methods Mol. Biol. 2015, 1241, 181–194.
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991.
- Chang, K.; Kang, P.; Liu, Y.; Huang, K.; Miao, T.; Sagona, A.P.; Nezis, I.P.; Bodmer, R.; Ocorr, K.; Bai, H. TGFB-INHB/activin signaling regulates age-dependent autophagy and cardiac health through inhibition of MTORC2. Autophagy 2020, 16, 1807–1822.
- Aspernig, H.; Heimbucher, T.; Qi, W.; Gangurde, D.; Curic, S.; Yan, Y.; Donner von Gromoff, E.; Baumeister, R.; Thien, A. Mitochondrial Perturbations Couple mTORC2 to Autophagy in C. elegans. Cell Rep. 2019, 29, 1399–1409.e5.
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 2019, 72, 51–62.
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141.
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461.
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. USA 2011, 108, 4788–4793.
- Bach, M.; Larance, M.; James, D.E.; Ramm, G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem. J. 2011, 440, 283–291.
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297.
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003.
- Wong, P.M.; Feng, Y.; Wang, J.; Shi, R.; Jiang, X. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun. 2015, 6, 8048.
- Torii, S.; Yoshida, T.; Arakawa, S.; Honda, S.; Nakanishi, A.; Shimizu, S. Identification of PPM1D as an essential Ulk1 phosphatase for genotoxic stress-induced autophagy. EMBO Rep. 2016, 17, 1552–1564.
- Park, J.M.; Seo, M.; Jung, C.H.; Grunwald, D.; Stone, M.; Otto, N.M.; Toso, E.; Ahn, Y.; Kyba, M.; Griffin, T.J.; et al. ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction. Autophagy 2018, 14, 584–597.
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750.
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell Biol. 2010, 12, 831–835.
- Xu, H.D.; Qin, Z.H. Beclin 1, Bcl-2 and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 109–126.
- Kuhlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89.
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol. 2019, 17, 53.
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta 2014, 1837, 418–426.
- Rimessi, A.; Previati, M.; Nigro, F.; Wieckowski, M.R.; Pinton, P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 2016, 81, 281–293.
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323.
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273.
- Perrone, M.; Caroccia, N.; Genovese, I.; Missiroli, S.; Modesti, L.; Pedriali, G.; Vezzani, B.; Vitto, V.A.M.; Antenori, M.; Lebiedzinska-Arciszewska, M.; et al. The role of mitochondria-associated membranes in cellular homeostasis and diseases. Int. Rev. Cell Mol. Biol. 2020, 350, 119–196.
- Patergnani, S.; Missiroli, S.; Marchi, S.; Giorgi, C. Mitochondria-Associated Endoplasmic Reticulum Membranes Microenvironment: Targeting Autophagic and Apoptotic Pathways in Cancer Therapy. Front. Oncol. 2015, 5, 173.
- Marchi, S.; Giorgi, C.; Galluzzi, L.; Pinton, P. Ca2+ Fluxes and Cancer. Mol. Cell 2020, 78, 1055–1069.
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730.
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554.
- Sato, K.; Sato, M. Multiple ways to prevent transmission of paternal mitochondrial DNA for maternal inheritance in animals. J. Biochem. 2017, 162, 247–253.
- Porter, G.A., Jr.; Hom, J.; Hoffman, D.; Quintanilla, R.; de Mesy Bentley, K.; Sheu, S.S. Bioenergetics, mitochondria, and cardiac myocyte differentiation. Prog. Pediatric Cardiol. 2011, 31, 75–81.
- Sin, J.; Andres, A.M.; Taylor, D.J.; Weston, T.; Hiraumi, Y.; Stotland, A.; Kim, B.J.; Huang, C.; Doran, K.S.; Gottlieb, R.A. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 2016, 12, 369–380.
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608.
- Ishihara-Paul, L.; Hulihan, M.M.; Kachergus, J.; Upmanyu, R.; Warren, L.; Amouri, R.; Elango, R.; Prinjha, R.K.; Soto, A.; Kefi, M.; et al. PINK1 mutations and parkinsonism. Neurology 2008, 71, 896–902.
- Klein, C.; Djarmati, A.; Hedrich, K.; Schafer, N.; Scaglione, C.; Marchese, R.; Kock, N.; Schule, B.; Hiller, A.; Lohnau, T.; et al. PINK1, Parkin, and DJ-1 mutations in Italian patients with early-onset parkinsonism. Eur. J. Hum. Genet. 2005, 13, 1086–1093.
- Hatano, Y.; Li, Y.; Sato, K.; Asakawa, S.; Yamamura, Y.; Tomiyama, H.; Yoshino, H.; Asahina, M.; Kobayashi, S.; Hassin-Baer, S.; et al. Novel PINK1 mutations in early-onset parkinsonism. Ann. Neurol. 2004, 56, 424–427.
- Ibanez, P.; Lesage, S.; Lohmann, E.; Thobois, S.; De Michele, G.; Borg, M.; Agid, Y.; Durr, A.; Brice, A.; French Parkinson’s Disease Genetics Study Group. Mutational analysis of the PINK1 gene in early-onset parkinsonism in Europe and North Africa. Brain J. Neurol. 2006, 129, 686–694.
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942.
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879.
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769.
- Hasson, S.A.; Kane, L.A.; Yamano, K.; Huang, C.H.; Sliter, D.A.; Buehler, E.; Wang, C.; Heman-Ackah, S.M.; Hessa, T.; Guha, R.; et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 2013, 504, 291–295.
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 2012, 22, 320–333.
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3, 1016.
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080.
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131.
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185.
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314.
- Yuan, Y.; Zheng, Y.; Zhang, X.; Chen, Y.; Wu, X.; Wu, J.; Shen, Z.; Jiang, L.; Wang, L.; Yang, W.; et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 2017, 13, 1754–1766.
- Shi, R.Y.; Zhu, S.H.; Li, V.; Gibson, S.B.; Xu, X.S.; Kong, J.M. BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. Ther. 2014, 20, 1045–1055.
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185.
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432.
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sorensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286.
- Lublin, F.D. New multiple sclerosis phenotypic classification. Eur. Neurol. 2014, 72 (Suppl. S1), 1–5.
- Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. J. Clin. Med. 2020, 9, 740.
- Alirezaei, M.; Fox, H.S.; Flynn, C.T.; Moore, C.S.; Hebb, A.L.; Frausto, R.F.; Bhan, V.; Kiosses, W.B.; Whitton, J.L.; Robertson, G.S.; et al. Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy 2009, 5, 152–158.
- Paunovic, V.; Petrovic, I.V.; Milenkovic, M.; Janjetovic, K.; Pravica, V.; Dujmovic, I.; Milosevic, E.; Martinovic, V.; Mesaros, S.; Drulovic, J.; et al. Autophagy-independent increase of ATG5 expression in T cells of multiple sclerosis patients. J. Neuroimmunol. 2018, 319, 100–105.
- Becher, J.; Simula, L.; Volpe, E.; Procaccini, C.; La Rocca, C.; D’Acunzo, P.; Cianfanelli, V.; Strappazzon, F.; Caruana, I.; Nazio, F.; et al. AMBRA1 Controls Regulatory T-Cell Differentiation and Homeostasis Upstream of the FOXO3-FOXP3 Axis. Dev. Cell 2018, 47, 592–607.e6.
- Akatsuka, H.; Kuga, S.; Masuhara, K.; Davaadorj, O.; Okada, C.; Iida, Y.; Okada, Y.; Fukunishi, N.; Suzuki, T.; Hosomichi, K.; et al. AMBRA1 is involved in T cell receptor-mediated metabolic reprogramming through an ATG7-independent pathway. Biochem. Biophys. Res. Commun. 2017, 491, 1098–1104.
- Astier, A.L. T-cell regulation by CD46 and its relevance in multiple sclerosis. Immunology 2008, 124, 149–154.
- Joubert, P.E.; Meiffren, G.; Gregoire, I.P.; Pontini, G.; Richetta, C.; Flacher, M.; Azocar, O.; Vidalain, P.O.; Vidal, M.; Lotteau, V.; et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009, 6, 354–366.
- Soldan, S.S.; Fogdell-Hahn, A.; Brennan, M.B.; Mittleman, B.B.; Ballerini, C.; Massacesi, L.; Seya, T.; McFarland, H.F.; Jacobson, S. Elevated serum and cerebrospinal fluid levels of soluble human herpesvirus type 6 cellular receptor, membrane cofactor protein, in patients with multiple sclerosis. Ann. Neurol. 2001, 50, 486–493.
- Xu, H.; Wu, Z.Y.; Fang, F.; Guo, L.; Chen, D.; Chen, J.X.; Stern, D.; Taylor, G.A.; Jiang, H.; Yan, S.S. Genetic deficiency of Irgm1 (LRG-47) suppresses induction of experimental autoimmune encephalomyelitis by promoting apoptosis of activated CD4+ T cells. FASEB J. 2010, 24, 1583–1592.
- Singh, S.B.; Ornatowski, W.; Vergne, I.; Naylor, J.; Delgado, M.; Roberts, E.; Ponpuak, M.; Master, S.; Pilli, M.; White, E.; et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat. Cell Biol. 2010, 12, 1154–1165.
- Guo, X.; Zhang, W.; Wang, C.; Zhang, B.; Li, R.; Zhang, L.; Zhao, K.; Li, Y.; Tian, L.; Li, B.; et al. IRGM promotes the PINK1-mediated mitophagy through the degradation of Mitofilin in SH-SY5Y cells. FASEB J. 2020, 34, 14768–14779.
- Bhattacharya, A.; Parillon, X.; Zeng, S.; Han, S.; Eissa, N.T. Deficiency of autophagy in dendritic cells protects against experimental autoimmune encephalomyelitis. J. Biol. Chem. 2014, 289, 26525–26532.
- Bonora, M.; De Marchi, E.; Patergnani, S.; Suski, J.M.; Celsi, F.; Bononi, A.; Giorgi, C.; Marchi, S.; Rimessi, A.; Duszynski, J.; et al. Tumor necrosis factor-alpha impairs oligodendroglial differentiation through a mitochondria-dependent process. Cell Death Differ. 2014, 21, 1198–1208.
- Holman, S.P.; Lobo, A.S.; Novorolsky, R.J.; Nichols, M.; Fiander, M.D.J.; Konda, P.; Kennedy, B.E.; Gujar, S.; Robertson, G.S. Neuronal mitochondrial calcium uniporter deficiency exacerbates axonal injury and suppresses remyelination in mice subjected to experimental autoimmune encephalomyelitis. Exp. Neurol. 2020, 333, 113430.
- Patergnani, S.; Castellazzi, M.; Bonora, M.; Marchi, S.; Casetta, I.; Pugliatti, M.; Giorgi, C.; Granieri, E.; Pinton, P. Autophagy and mitophagy elements are increased in body fluids of multiple sclerosis-affected individuals. J. Neurol. Neurosurg. Psychiatry 2018, 89, 439–441.
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Fainardi, E.; Casetta, I.; Granieri, E.; Pugliatti, M.; Pinton, P. Correlation between auto/mitophagic processes and magnetic resonance imaging activity in multiple sclerosis patients. J. Neuroinflamm. 2019, 16, 131.
- Albanese, M.; Zagaglia, S.; Landi, D.; Boffa, L.; Nicoletti, C.G.; Marciani, M.G.; Mandolesi, G.; Marfia, G.A.; Buttari, F.; Mori, F.; et al. Cerebrospinal fluid lactate is associated with multiple sclerosis disease progression. J. Neuroinflamm. 2016, 13, 36.
- Joodi Khanghah, O.; Nourazarian, A.; Khaki-Khatibi, F.; Nikanfar, M.; Laghousi, D.; Vatankhah, A.M.; Moharami, S. Evaluation of the Diagnostic and Predictive Value of Serum Levels of ANT1, ATG5, and Parkin in Multiple Sclerosis. Clin. Neurol. Neurosurg. 2020, 197, 106197.
- Hassanpour, M.; Cheraghi, O.; Laghusi, D.; Nouri, M.; Panahi, Y. The relationship between ANT1 and NFL with autophagy and mitophagy markers in patients with multiple sclerosis. J. Clin. Neurosci. 2020, 78, 307–312.
- Harberts, E.; Datta, D.; Chen, S.; Wohler, J.E.; Oh, U.; Jacobson, S. Translocator protein 18 kDa (TSPO) expression in multiple sclerosis patients. J. Neuroimmune Pharmacol. 2013, 8, 51–57.
- Nohara, S.; Ishii, A.; Yamamoto, F.; Yanagiha, K.; Moriyama, T.; Tozaka, N.; Miyake, Z.; Yatsuga, S.; Koga, Y.; Hosaka, T.; et al. GDF-15, a mitochondrial disease biomarker, is associated with the severity of multiple sclerosis. J. Neurol. Sci. 2019, 405, 116429.
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32.
- Vezzani, B.; Carinci, M.; Patergnani, S.; Pasquin, M.P.; Guarino, A.; Aziz, N.; Pinton, P.; Simonato, M.; Giorgi, C. The Dichotomous Role of Inflammation in the CNS: A Mitochondrial Point of View. Biomolecules 2020, 10, 1437.
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522.
- Mark, R.J.; Pang, Z.; Geddes, J.W.; Uchida, K.; Mattson, M.P. Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: Involvement of membrane lipid peroxidation. J. Neurosci. 1997, 17, 1046–1054.
- De Wilde, M.C.; Vellas, B.; Girault, E.; Yavuz, A.C.; Sijben, J.W. Lower brain and blood nutrient status in Alzheimer’s disease: Results from meta-analyses. Alzheimer’s Dement. 2017, 3, 416–431.
- Luth, H.J.; Munch, G.; Arendt, T. Aberrant expression of NOS isoforms in Alzheimer’s disease is structurally related to nitrotyrosine formation. Brain Res. 2002, 953, 135–143.
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166.
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122.
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Abeta secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69.
- Piras, A.; Collin, L.; Gruninger, F.; Graff, C.; Ronnback, A. Autophagic and lysosomal defects in human tauopathies: Analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. Commun. 2016, 4, 22.
- Inoue, K.; Rispoli, J.; Kaphzan, H.; Klann, E.; Chen, E.I.; Kim, J.; Komatsu, M.; Abeliovich, A. Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol. Neurodegener. 2012, 7, 48.
- Kesharwani, R.; Sarmah, D.; Kaur, H.; Mounika, L.; Verma, G.; Pabbala, V.; Kotian, V.; Kalia, K.; Borah, A.; Dave, K.R.; et al. Interplay between Mitophagy and Inflammasomes in Neurological Disorders. ACS Chem. Neurosci. 2019, 10, 2195–2208.
- Martin-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; Garcia-Escudero, V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 792–806.
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412.
- Schwagerl, A.L.; Mohan, P.S.; Cataldo, A.M.; Vonsattel, J.P.; Kowall, N.W.; Nixon, R.A. Elevated levels of the endosomal-lysosomal proteinase cathepsin D in cerebrospinal fluid in Alzheimer disease. J. Neurochem. 1995, 64, 443–446.
- Sundelof, J.; Sundstrom, J.; Hansson, O.; Eriksdotter-Jonhagen, M.; Giedraitis, V.; Larsson, A.; Degerman-Gunnarsson, M.; Ingelsson, M.; Minthon, L.; Blennow, K.; et al. Higher cathepsin B levels in plasma in Alzheimer’s disease compared to healthy controls. J. Alzheimer’s Dis. 2010, 22, 1223–1230.
- Armstrong, A.; Mattsson, N.; Appelqvist, H.; Janefjord, C.; Sandin, L.; Agholme, L.; Olsson, B.; Svensson, S.; Blennow, K.; Zetterberg, H.; et al. Lysosomal network proteins as potential novel CSF biomarkers for Alzheimer’s disease. Neuromol. Med. 2014, 16, 150–160.
- Sjodin, S.; Brinkmalm, G.; Ohrfelt, A.; Parnetti, L.; Paciotti, S.; Hansson, O.; Hardy, J.; Blennow, K.; Zetterberg, H.; Brinkmalm, A. Endo-lysosomal proteins and ubiquitin CSF concentrations in Alzheimer’s and Parkinson’s disease. Alzheimer’s Res. Ther. 2019, 11, 82.
- Francois, A.; Julian, A.; Ragot, S.; Dugast, E.; Blanchard, L.; Brishoual, S.; Terro, F.; Chassaing, D.; Page, G.; Paccalin, M. Inflammatory Stress on Autophagy in Peripheral Blood Mononuclear Cells from Patients with Alzheimer’s Disease during 24 Months of Follow-Up. PLoS ONE 2015, 10, e0138326.
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Bosi, C.; Brombo, G.; Pugliatti, M.; Seripa, D.; Zuliani, G.; et al. Autophagy and mitophagy biomarkers are reduced in sera of patients with Alzheimer’s disease and mild cognitive impairment. Sci. Rep. 2019, 9, 20009.

