 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tao Jin | + 6933 word(s) | 6933 | 2021-04-12 04:58:20 | | | |
| 2 | Conner Chen | Meta information modification | 6933 | 2021-04-12 11:58:34 | | | | |
| 3 | Conner Chen | Meta information modification | 6933 | 2021-04-12 12:02:07 | | | | |
| 4 | Conner Chen | Meta information modification | 6933 | 2021-05-10 05:09:46 | | | | |
| 5 | Conner Chen | Meta information modification | 6933 | 2021-05-10 05:10:23 | | |
Video Upload Options
Staphylococcus aureus (S. aureus) infections are a major healthcare challenge and new treatment alternatives are needed. S. aureus septic arthritis, a debilitating joint disease, causes permanent joint dysfunction in almost 50% of the patients. S. aureus bacteremia is associated with higher mortalities than bacteremia caused by most other microbes and can develop to severe sepsis and death. The key to new therapies is understanding the interplay between bacterial virulence factors and host immune response, which decides the disease outcome. S. aureus produces numerous virulence factors that facilitate bacterial dissemination, invasion into joint cavity, and cause septic arthritis. Monocytes, activated by several components of S. aureus such as lipoproteins, are responsible for bone destructions. In S. aureus sepsis, cytokine storm induced by S. aureus components leads to the hyperinflammatory status, DIC, multiple organ failure, and later death.
1. S. aureus and Its Virulence Factors
Nearly half of the human population is at some point colonized by Staphylococcus aureus (S. aureus). Of these, 20% are persistently colonized while around 30% are intermittently colonized, mostly in the anterior nares and the skin [1]. However, one should not make the mistake of assuming that S. aureus is a harmless microbe that is only part of the normal flora. Rather, S. aureus is indeed a very virulent bacteria that causes a wide range of diseases, from simple wound infections and food poisoning to life-threatening conditions such as sepsis, meningitis, and endocarditis [2]. In this review, we focus on septic arthritis and sepsis, which are two of the many severe infections caused by S. aureus.
S. aureus is a very resilient pathogen due to the various virulence factors it contains and produces, some of which are described below and illustrated in Figure 1. The biological function of S. aureus virulence factors and their roles in infections are summarized in Table 1, reviewed in [3].
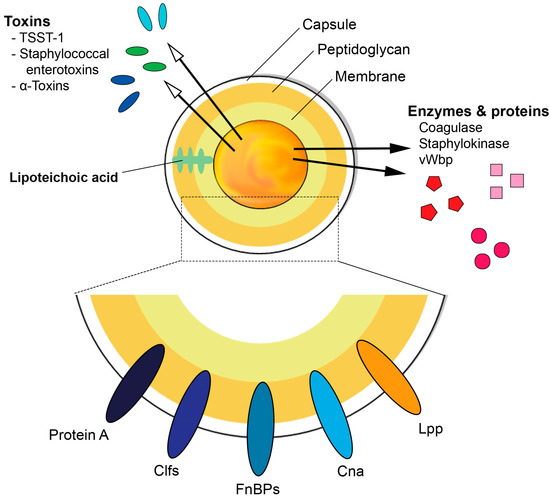
Figure 1. Schematic diagram illustrating the basic structure of Staphylococcus aureus and its ability to express various virulence factors. TSST-1 = Toxic shock syndrome toxin-1, Clfs = clumping factors, FnBPs = fibronectin binding proteins, Cna = Collagen adhesin, Lpp = lipoproteins, vWbp = von Willebrand factor-binding protein.
Table 1. S. aureus virulence factors and their roles in infections.
| Biological Functions | Role in Infection | |
|---|---|---|
| Cell Wall Components: | ||
| Capsular Polysaccharide | Antiphagocytic [4] | Septic arthritis, sepsis [5] |
| Peptidoglycan | Release of TNF-α, IL-6 [6] | Arthritis [6], bone loss [7] |
| Lipoproteins | Activation of innate immunity [8] | Arthritis [9], sepsis [10] |
| Surface Proteins: | ||
| ClfA | Inhibit complement-mediated phagocytosis [11][12] | Septic arthritis, sepsis [13] |
| SpA | Inhibit opsonophagocytosis, B-cell superantigen [14] | Septic arthritis, sepsis [15] |
| FnBPs | Adhesion and invasion of cells [11] | Biofilm formation [11], sepsis [16] |
| Cna | Mediates binding of S. aureus to cartilage [17] | Septic arthritis [18] |
| Secreted Proteins: | ||
| vWbp | Promote blood-clotting [19] | Septic arthritis [20] |
| Eap | Adhesin [21][22] | Inhibit wound healing, biofilm formation [21][22] |
| Staphylokinase | Mediates digestion of fibrin clots [23] | S. aureus skin infections [23] |
| Toxins: | ||
| TSST-1 | Superantigen [24] | Toxic shock syndrome [25], septic arthritis [26] |
| SE | Superantigen [24] | Toxic shock syndrome [27], food poisoning [28] |
| SEls | Superantigen [24] | Food poisoning [28] |
| α- and γ-toxins | Cell lysis [29] | Septic arthritis [30] |
| PVL | Cell lysis [29] | Necrotizing pneumonia [31] |
| PSMs | Cell lysis [29], induce inflammation [32] | Biofilm formation [32][33][34], osteomyelitis [35] |
| Bacterial DNA | Release of TNF-α, IL-6, IFN-γ [36] | Arthritis [37], septic shock [38] |
Abbreviations: ClfA = Clumping factor A; SpA = Staphylococcal protein A; FnBPs = Fibronectin binding proteins; Cna = Collagen adhesion; vWbp = von Willebrand factor binding protein; Eap = extracellular adherence protein; TSST-1 = Toxic shock syndrome toxin 1; SE = Staphylococcal enterotoxins; SEls = Staphylococcal enterotoxin like toxins; PVL = Panton-Valentine leucocidin; PSMs = Phenol soluble modulins.
1.1. Cell Wall Components
S. aureus expresses a capsular polysaccharide (CP) that functions as a virulent factor, enabling the bacteria to evade phagocytosis [4]. Several serotypes of the CP have been identified and of those, CP5 and CP8 are the major ones. Most of the clinical isolates of S. aureus have the capability to produce either CP5 or CP8 [4]. S. aureus strains expressing the CP5 capsule significantly increase mortality and arthritis frequency and severity in S. aureus induced sepsis and septic arthritis, respectively, compared to the strains lacking the CP5 capsule [5]. This can be due to the downregulatory effects of CP5 on the uptake and intracellular killing ability of the phagocytes [5]. The CP8 serotype seems to be less virulent than the CP5 serotype as demonstrated by the ability of CP5 to cause higher bacteremia than the CP8 serotype in a mouse model of bacteremia. The CP5 producing strain also exhibited greater resistance to in vitro opsonophagocytic killing by neutrophils compared to the CP8 serotype [39].
The cell wall of S. aureus is made up of a 20–30 nm thick layer of peptidoglycan (PGN). Apart from being a protective barrier of the bacteria, PGN has other functions such as being a scaffold, whereby surface proteins that are fundamental for bacterial virulence can attach [40]. The major structural features of PGN consist of linear glycan strands made up of alternating N-acetylglucosamine and N-acetylmuramic acid residues that are linked by β-1-4 bonds [41]. The glycan strands are cross-linked by short peptides made up of D-alanine, L-lysine, D-glutamic acid, and L-alanine [42]. The ɛ-amino groups L-lysine of nearby peptides are cross-linked to D-alanine of other peptides through pentaglycine bridges, thus giving rise to the 3-dimensional structure of the PGN [43]. Due to the critical role it plays in maintaining bacterial structure, growth, and viability, PGN is a target for antibiotics and for the immune system. Nod-like receptors (NLRs), mannose-binding lectin, and lysozyme are some of the components of the immune system that can recognize and target PGN [44]. PGN is a strong inducer of inflammation and stimulates the release of proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β, and IL-6. Furthermore, studies have shown that PGN alone can induce arthritis in mice [6] and repetitive inhalation of PGN components can lead to bone loss [7]. However, compared to an extremely potent effect of S. aureus lipoproteins (Lpps) the proinflammatory capacity of PGN is mild and transient [9].
Besides, secondary modifications of the PGN cell wall are of vital importance in order to resist host immune response enforcements [45]. Antibacterial enzymes produced by the host, such as lysozyme, also known as muramidase, can cleave PGN in the β-1,4 linkage site localized between the sugar residues of N-acetylglucosamine and N-acetylmuramic acid, thereby inhibiting bacterial overgrowth [45]. However, S. aureus impressively protects itself from “cell wall breakdowns” by utilizing O-acetylation modifications mediated by the O-acetyltransferase A (OatA) enzyme in the PGN cell wall [46]. We demonstrated that the virulence associated with PGN OatA, in both systemic and local S. aureus-induced septic arthritis model, led to milder progression of the disease in mice infected with a ΔoatA mutant strain [47], indicating the pathogenic importance of the O-acetylation of PGN in staphylococcal septic arthritis.
Among the constituents of the cell wall of S. aureus are the teichoic acids, made up of polyribitol and polyglycerol phosphates and sugar-containing polymers [48][49]. Teichoic acids are not unique to S. aureus but are found in various Gram-positive bacteria. They can be categorized into wall teichoic acids (WTAs) that are covalently attached to the bacterial PGN, and lipoteichoic acids (LTAs) that are anchored to the lipid membrane through a glycolipid [48][49]. Historically, LTA has been regarded as a potent stimulator of the innate immunity upon its recognition by Toll like receptor 2 (TLR2) among others, leading to the activation of the macrophages and the release of proinflammatory cytokines [50][51]. However, recent studies cast a shadow of doubt on these results, pointing a finger to contamination by S. aureus Lpps.
S. aureus Lpp consists of a lipid-moiety and a protein-moiety. The lipid portion is covalently attached to a cysteine residue in the N-terminal region, ultimately facilitating its anchoring in the outer leaflet of the bacterial cytoplasmic membrane [52]. Lpp in S. aureus plays an essential role in enabling the bacteria to acquire sufficient loads of iron under infectious conditions. As free iron ions are limited in the host environment and the iron supply is of critical importance for the survival of the staphylococcal pathogen [53][54], inhibition of the Lpp function, through incomplete Lpp maturation, can be deleterious to the metabolic fitness of the bacterium, consequently prompting it to cease in the battle against the host. The lipid structures of Lpp in S. aureus are known as microbe-associated molecular patterns (MAMPs) [52], being vital activators of the innate immunity as they play a potent role in alerting TLR2 in host cells [8].
S. aureus Lpp behave differently in different murine models, depending on the route of infection, the organ of interest, and the assessed time points. In a local knee arthritis model, S. aureus Lpps mediated severe arthritogenic and bone destructive effects in NMRI and C57BL/6 wild-type mice, but not in TLR2 deficient mice after intra-articular knee joint challenge with the purified Lpp. The arthritogenic properties were mediated through the lipid-moiety [9]. Interestingly, S. aureus SA113 Δlgt mutant strain that lacks Lpp maturation, induced more knee swelling compared to its parental strain. This coincided with increased IL-6 expression and higher bacterial burden in the local knee [9]. In contrast, in a hematogenous septic arthritis model, increased bacterial persistence was observed in both C57BL/6 wild-type and TLR2 deficient murine kidneys when inoculated intravenously with the S. aureus Newman parental strain compared to its Δlgt mutant strain [55]. This is in agreement with earlier reports showing that different organs, including the kidneys, exhibited higher bacterial loads when infected with SA113 parental strain in comparison to the Δlgt mutant strain, independent of TLR2 and MyD88 signaling [10]. In addition, USA300 MRSA parental strain led to higher bacterial burden in kidneys of Balb/c mice compared to its Δlpl mutant strain, lacking the lpl gene cluster [56].
1.2. Surface Proteins
S. aureus expresses several surface proteins that play a crucial role in enabling the bacteria to adhere to the host cells, aid in invasion of the bacteria, and evade the immune response mounted by the host [11]. Adherence of bacterial products to host tissues is one of the important steps in initiation of colonization and infections [57]. S. aureus surface proteins usually recognize and adhere to several components of the extracellular matrix (ECM) such as fibronectin, fibrin, and collagen [57].
Microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) are S. aureus surface proteins that are anchored on the cell wall PGN. Most MSCRAMMs contain a carboxyl-terminal sorting signal containing LPXTG motif that is cleaved by S. aureus sortase enzymes before being covalently anchored to the cell wall PGN [58]. Notable members of this group include fibronectin binding protein A and B (FnBPA and FnBPB), collagen adhesin (Cna), staphylococcal protein A (SpA), and clumping factor A and B (ClfA and ClfB) [11].
ClfA binds to soluble fibrinogen and has been shown to inhibit complement-mediated phagocytosis [11][12]. In S. aureus induced septic arthritis and sepsis, ClfA is an important virulence factor that promotes the pathogenesis of the diseases and can be a target for generation of vaccine against S. aureus infections. Passive immunization of mice with rat and rabbit anti-ClfA antibodies gave protection against S. aureus induced septic arthritis and sepsis [13].
SpA not only evades the innate immunity by inhibiting opsonophagocytosis, but also has the ability to alter the response of the adoptive immunity by binding to both Fc region of IgG and Fab regions of the B-cell receptor, thereby inducing apoptosis by functioning as a B-cell superantigen [14]. SpA can also diminish the proinflammatory signaling of TNF-α by binding to its receptor, tumor necrosis factor receptor 1 (TNFR1) [59].
FnBPs are important in helping the bacteria to adhere to and invade cells of the host and together with ClfB and SpA play a role, although not yet fully understood, in forming S. aureus biofilms [11]. FnBPs are also involved in the pathogenesis of S. aureus induced sepsis [16].
Cna binds to collagen and helps to mediate the binding of S. aureus to cartilage [17] and was shown by Patti et al. to be involved in the pathogenesis of S. aureus septic arthritis [18]. Mice injected with S. aureus lacking Cna developed significantly less frequent signs of clinical arthritis (27%) compared with mice infected with wild type S. aureus (70%) [18].
S. aureus also secretes other proteins that, although not covalently attached to the cell wall PGN, are still surface-associated proteins and act as adhesins—they are commonly known as secreted expanded repertoire adhesive molecules (SERAMs). Coagulase (Coa), von Willebrand factor binding protein (vWbp), extracellular fibrinogen binding protein and extracellular adherence protein (Eap) are several examples of SERAMs [60]. The ability of S. aureus to clot human blood is mediated by the direct binding of Coa and vWbp with the hosts’ prothrombin. The resulting staphylothrombin complex will eventually convert fibrinogen to fibrin, thus forming fibrin clots [19]. Additionally, as its name suggests, vWbp acts as a bridge between the bacterial cell wall and von Willebrand factor (vWf) therefore enabling adhesion of S. aureus to vascular tissues [61][62]. Intriguingly, vWbp but not Coa expressed by S. aureus facilitates the initiation of septic arthritis and such an effect might be mediated through its interaction with a host factor (von Willebrand factor), strongly suggesting bacterial adherence to blood vessels is more important than fibrin clotting function of coagulases in induction of septic arthritis [20]. Eap plays multiple roles in S. aureus infections such as acting as an adhesin, inhibiting wound healing and being involved in biofilm formation [21][22].
1.3. Secreted Proteins
S. aureus superantigen like proteins (SSLs) are another group of proteins secreted by S. aureus that have similar structures as superantigens but lack superantigenic activities. Several SSLs have been identified that are able to interfere with the innate immune response [22][63]. Of these, SSL3 can bind to TLR2 and inhibit the production of TNF-α by macrophages stimulated by heat-killed S. aureus or PGN [22][63].
S. aureus also secretes numerous other proteins such as the chemotaxis inhibitory protein of S. aureus, staphylococcal complement inhibitor, and formyl peptide receptor-like-1 inhibitory protein that aid the bacteria to evade opsonization and phagocytosis [2].
Enzymes secreted by S. aureus include catalase, proteases, hyaluronidase, lipases, nucleases, and staphylokinase. Apart from exploiting host tissues and converting them into nutrients for the bacteria, S. aureus enzymes also facilitate invasion and evasion of the immune system [2]. Hyaluronidase breaks down hyaluronic acid that holds the cells of the body together, thus facilitating the invasion of S. aureus further into tissues [64]. Staphylokinase, which mediates the digestion of fibrin clots via activation of plasminogen to plasmin, has been shown to promote the establishment of S. aureus skin infections, but at the same time decrease the severity of the disease [23]. Intriguingly, fibrinolysis activated by staphylokinase prevents biofilm formation and promotes detachment of biofilms [65].
1.4. Toxins
The secretion of toxins is another virulence weapon of S. aureus that bacteria use to manipulate and gain the upper hand against the immune system. S. aureus secretes large amounts of toxins with several different virulence factors. Several toxins secreted by S. aureus have superantigenic properties, i.e., the ability to cause non-specific activation of T-cells, leading to massive polyclonal T cell activation followed by a vast release of cytokines with subsequent fever, shock, and multiple organ failure [66][67]. It was long assumed that superantigens bind only to the T cell receptor (TCR) on the T-cells and major histocompatibility complex (MHC) class II molecules on antigen presenting cells (APCs) [68]. However, it has since emerged that superantigens can also bind to CD28, thus forming a more stable complex than previously thought [69].
Toxic shock syndrome toxin 1 (TSST-1), staphylococcal enterotoxins (SEs) (AE, G-1, R and T), and staphylococcal enterotoxin like toxins (SEls) (J-Q, S, U, V, and X) are all superantigenic toxins produced by S. aureus [24]. Of the SEs, SEB and SEC are known to cause non-menstrual toxic shock syndrome (TSS) [27]. Furthermore, SEs have long been known to cause food poisoning whereas SEls were thought not to have emetic properties. However, recent studies have found that some newly discovered SEls (I-Q) have emetic properties and may also play some role in staphylococcal food poisoning [28]. TSST-1 accounts for almost half of all non-menstrual TSS in the general population and almost all cases of menstruation associated TSS [25]. In addition, clonal expansion of CD4+ Vβ11+ T cells induced by S. aureus producing TSST-1 toxin has been shown to be involved in the pathogenesis of S. aureus septic arthritis [26].
Another set of toxins secreted by S. aureus includes the hemolysins (also known as alpha (α), beta (β), and gamma (γ) toxins), cytolytic peptides (phenol soluble modulins (PSMs)) and bi-component leukocidins (including Panton-Valentine leukocidin (PVL)). All are characterized by their ability to cause cell lysis by forming pores in the cell membrane [29].
The first to be discovered and most studied of the hemolysins is the α-toxin, with an ability to form pores and lyse a broad range of cell types such as peripheral blood monocytes, platelets, and keratinocytes, and cells of the endothelium [70]. In addition to α toxins, γ toxins produced by S. aureus are also a critical virulence factor in S. aureus induced septic arthritis since mice injected with a mutant lacking both α and γ toxins showed significantly less frequent and less severe arthritis compared to wild-type strains producing both α and γ toxins [30].
As mentioned, PVL has adhesion properties, damages leukocytes and has also been implicated in the pathogenesis of necrotizing pneumonia [31].
The PSMs consist of small peptides and similar to the hemolysins and leukocidins have pore-forming properties. The PSMs can target several cell types such as erythrocytes and leukocytes and have been shown to induce inflammation [32]. Members of this family include the PSM-mec, PSMα 1-4, PSMβ 1-2, and PSMγ [32]. Apart from the role in biofilm structuring and dispersal [32][33][34], PSMs facilitate invasion and killing of osteoblasts thereby aggravating S. aureus-induced osteomyelitis [35]. PSMs are largely produced by community-associated methicillin-resistant S. aureus (CA-MRSA). Furthermore, they are involved in biofilm formation and are known to cause aggressive S. aureus infections [34].
1.5. Bacterial DNA
Bacterial but not mammalian DNA can induce inflammatory response. The proinflammatory properties of bacterial DNA are largely dependent on bacterial CpG motifs that are unmethylated cytosine–phosphate–guanine (CpG) dinucleotides. The CpG motifs are predominantly prevalent only in bacterial DNA, but not in mammalian DNA [71]. S. aureus DNA can induce the production of proinflammatory cytokines such as TNF-α, IL-6, and interferon-gamma (IFN-γ) via TLR9 [36]. Indeed, the injection of S. aureus DNA in mice led to rapid activation of macrophages followed by massive release of TNF-α that triggered lethal shock in mice [38]. Furthermore, previous results showed that S. aureus DNA containing CpG motifs induced arthritis [37] and meningitis through NF-κB [72]. However, our recent data suggest that DNA from antibiotic-killed S. aureus plays a minor role in mediating arthritis [73].
2. The Immune Response during S. aureus Infections
S. aureus, through its vast virulence factors, seeks multiple ways to colonize and establish infections in humans. However, upon intrusion, this pathogenic bacterium highly alerts the host’s immune system. Consequently, a battle between the host and the pathogen starts. The role of different immune cells in S. aureus septic arthritis and sepsis is summarized in Table 2.
Table 2. The role of different immune cells in S. aureus septic arthritis and sepsis.
| Cell Types | Septic Arthritis | Bacterial Clearance | Sepsis |
|---|---|---|---|
| Neutrophils | Protective [74] | Enhance [74] | Pathogenic [74] |
| Monocytes/macrophages | Pathogenic [75] | Enhance [75] | Protective [75] |
| NK cells | Protective [76] | Enhance [76] | Protective [76] |
| Innate lymphoid cells | NA | NA | Type 2 ILCs are protective [77] |
| CD4+ T cells | Pathogenic [78] | No effect [78] | Pathogenic [78] |
| CD8 T cells | NA | NA | NA |
| Regulatory T cells | Protective [79] | No effect [79] | NA |
| Th17 T cells | NA | NA | NA |
| B cells | No effect [80] | No effect [80] | Protective? [80] |
| Thrombocytes | NA | NA | Protective [81] |
| Eosinophils | NA | NA | Protective [77] |
| Basophils | NA | NA | Protective [82] |
NA = not assessed; ILCs = innate lymphoid cells.
2.1. Innate Immunity
The host’s innate immune system immediately executes a series of protective measures against intruding pathogens, such as S. aureus, and this serves as the first line of defense [83]. This action is initially implemented through recognition via pattern recognition receptors (PRRs) that distinctively sense pathogenic components and promptly trigger the activation of innate immune cells [83]. Among these immune cells are the phagocytes.
2.2. Neutrophils
Neutrophils are the most abundant type of white blood cells (WBCs) in the body, constituting around 50–70% of all WBCs and play a very important role in the innate immunity. During S. aureus infections, neutrophils are quickly recruited from the blood and migrate to the infection site via a process known as chemotaxis [84]. Neutrophils have several PRRs, such as Toll like receptors (TLRs) that can recognize different conserved molecules from microbes, so called pathogen-associated molecular patterns (PAMPs). At the infection site, PRRs on the neutrophils will recognize PAMPs from bacteria, which is subsequently internalized. Around the internalized bacteria, a cellular compartment known as phagosome will be formed, which fuses with lysosomes to form phagolysosomes. The rapid release of reactive oxygen species through oxidative burst, antibacterial peptides that have microbicidal effects, proteinases that degrade bacterial components, and proteins that sequester essential bacterial nutrients are some of the mechanisms employed by the neutrophils in the phagolysosomes to neutralize the internalized bacteria [85].
Neutrophils also possess the ability to kill bacteria extracellularly by releasing its contents and DNA, known as neutrophil extracellular traps (NETs). This process involves forming a web-like structure interconnected with histones and containing antimicrobial agents such as defensins and myeloperoxidase that trap the bacteria and eliminate it [86]. Neutrophils are killing machines that do an excellent job phagocytizing bacteria and thus have a short life span (1–2 days) as a regulatory precaution to avoid tissue damage. Neutrophils are absolutely essential in protecting the host against live S. aureus infections, as clearly exhibited by the significantly higher mortality and more severe arthritis caused by S. aureus in neutrophil-depleted mice compared to wild-type controls [74]. In addition, in the murine S. aureus skin infection model, Mölne et al. demonstrated that neutrophil depletion worsens the disease severity with increased bacterial burden in the skin tissue [87]. On the other hand, the depletion of neutrophils did not have any impact in arthritis caused either by antibiotic-killed S. aureus or S. aureus Lpp [9][73].
2.3. Macrophages
Macrophages are outstanding phagocytes that not only eliminate S. aureus but also function as APCs and are involved in activating the adaptive immunity in case of serious breaches. Activated macrophages are also potent secretors of the proinflammatory cytokine TNF-α, whose role in S. aureus infections will be described later.
Two distinct subtypes of macrophages have been described with opposing activities: M1 macrophages and M2 macrophages. M1 macrophages, also known as “classically activated” macrophages, are proinflammatory. The enzyme nitric oxide synthase (iNOS) is expressed by M1 macrophages and helps convert arginine into nitric oxide (NO), which inhibits proliferation of infected cells [88][89]. Microbial products, such as lipopolysaccharides (LPS) or the proinflammatory cytokine IFN-γ, stimulate the M1 macrophages phenotype that will result in a Th1 immune response [90]. This will lead to the production of more proinflammatory cytokines such as IL-12, TNF-α, and IFN-γ in a positive feedback loop, thus maintaining the M1 macrophage phenotype. M2 macrophages, or “alternatively activated macrophages”, are anti-inflammatory and give rise to a Th2 immune response and thus promote cell proliferation and wound repair. The anti-inflammatory cytokine IL-4 promotes the differentiation of macrophages into M2 macrophages and stimulates the production of IL-10, which further enhances the phenotype of M2 macrophages [91].
Macrophages have specific names depending on the tissue on which they reside. For example, Kupffer cells are macrophages that are found in the liver, whereas microglia, adipose tissue macrophages, and osteoclasts are found in the central nervous system, adipose tissue, and bones, respectively [92]. Osteoclasts and osteoblasts play an important role in maintaining bone homeostasis by degrading and synthesizing bones, respectively [93]. IL-15, a proinflammatory pleiotropic cytokine, plays a potent role in early osteoclast differentiation [94]. Additionally, the receptor activator of nuclear factor kappa-B-ligand (RANKL)-dependent osteoclastogenesis is known to be impaired in the mice lacking the IL-15 receptor [95]. In S. aureus septic arthritis, mice lacking IL-15 were found to have a reduced number of osteoclasts in their joints, which also coincided with reduced arthritis severity and less joint destruction compared to wild-type mice [96]. Activation of osteoclasts requires the RANKL, a member of the tumor necrosis factor (TNF) superfamily that is found on the surface of osteoblasts, to bind to receptor activator of nuclear factor kappa-B (RANK) on the surface of osteoclasts. RANKL has been implicated in S. aureus infections, and its inhibition reduces bone loss in S. aureus septic arthritis [97].
Macrophages have been shown to play dual roles in S. aureus infections. On the one hand, Verdrengh et al. showed that macrophages are involved in aggravating S. aureus arthritis and the deficiency of macrophages attenuated the disease [75]. On the other hand, the ability of the host to clear invading bacteria in the kidneys is impeded, thus leading to higher mortality [75]. Further studies also showed that macrophages are involved in arthritis triggered by bacterial DNA containing CpG motifs [98]. We recently demonstrated that purified S. aureus Lpp rapidly initiates the recruitment of monocytes/macrophages and neutrophils upon local knee injection [9]. In the model of local knee arthritis induced by purified S. aureus Lpp, depletion of monocytes/macrophages resulted in diminished bone destruction [9]. This demonstrates that monocytes/macrophages are the key cell types in the development of local knee arthritis induced by purified Lpp. Similarly, double depletion of both neutrophils and monocytes abrogated the arthritis induced by antibiotic-killed S. aureus [73].
2.4. Natural Killer (NK) Cells
NK cells are a type of white blood cells that play an important role in the innate immune system. NK cells respond to and eliminate virus-infected and tumor cells and do not require antibodies or MHC to respond to these cells. NK cells play a protective role during S. aureus infections [76][99]. The depletion of NK cells in mice before inoculation with a TSST-1 secreting strain of S. aureus is associated with higher susceptibility to develop S. aureus septic arthritis as compared to control mice with intact NK cells [76]. Further studies have also shown that mice depleted of NK cells are significantly more susceptible to pulmonary S. aureus infections compared to wild-type mice [99], underscoring the protective role of NK cells against S. aureus infections.
2.5. The Complement System
The complement system serves as the first line of defense and is a crucial part of the innate immune system. It is made up of several plasma proteins and can be activated through three different pathways: the classical, the alternative and the lectin pathway. Whenever bacteria are successful in breaching the physical barriers, the complement system, regardless of the activation pathway, will recognize this and form enzyme complexes known as C3 convertases whose task is to cleave the complement component 3 (C3) into two different proteins. The C3a, a proinflammatory anaphylatoxin, helps with the recruitment of the phagocytes to the infection site, whereas the C3b opsonizes the invading S. aureus, thus making it easier to be phagocytosed [100].
Apart from opsonizing the bacteria, the complement system can also form a lytic complex known as the membrane attack complex (MAC) on the surface of invading bacterial cells that will lead to the lysis and eventual death of the microbe. However, the MAC recognizes only Gram-negative bacteria and thus S. aureus is spared from the potent killing ability of the MAC mechanism [101].
The complement system is imperative to the host defense during S. aureus infection as its deficiency renders the host defenseless and significantly increases the susceptibility to S. aureus infections [102]. Recent data from our lab show that mice lacking the complement component 3 (C3-/-) are highly susceptible to S. aureus septic arthritis. Kidney abscess formation and bacterial burden in the kidneys are also negatively affected in the C3-/- mice compared to the wild type controls with functioning complement system [103]. The results underscore the importance of the complement system in fending off S. aureus infections.
2.6. Adaptive Immunity
2.6.1. T-Cells
T-cells or T-lymphocytes are an integral part of adaptive immunity. They originate in the bone marrow but mature in the thymus, hence the name T-cells. T-cells are recognized from other lymphocytes due to their unique TCR displayed on the cell surface.
T-helper (Th) cells (CD4+ T-cells) express CD4 glycoprotein on their surface, recognize antigens presented by MHC class II and secrete cytokines that are necessary for both the cell-mediated and humoral immune response [104]. Cytotoxic T-cells (CD8+ T-cells) express CD8 glycoproteins, recognize antigens presented by MHC class I and eliminate virus-infected and tumor cells [104]. Regulatory T-cells (Tregs) play an important role in maintaining balance by preventing the immune response to self-antigens and suppressing excessive immune response that can cause autoimmune diseases [105]. T-helper 17 (Th17) cells are a unique CD4+ T-cell subset characterized by the production of IL-17 that is a highly inflammatory cytokine playing an important role in the pathogenesis of several autoimmune diseases [106].
CD4+ T-cells differentiate into two major subgroups: Th1 and Th2 cells. Th1 cells mainly secrete the cytokines IFN-γ and IL-2, respond to intracellular microbes and stimulate phagocyte mediated uptake and elimination of microbes [107][108]. Th2 cells usually respond to extracellular pathogens such as gastrointestinal parasites, secrete mainly IL-4 and IL-5 cytokines and promote eosinophil activation and phagocyte-independent immune response [107]. Cytotoxic T-lymphocyte-associated protein 4 (CTLA4), a naturally occurring protein receptor expressed on the surface of the T-cells, has the ability to inhibit the activation of the T-cell by competitively binding to CD80/86. CTLA4-Ig, a biologic that inhibits the full activation of T-cells, downregulates the Th2 response, and has little effect on Th1 response [109]. Septic arthritis mice pretreated with CTLA4-Ig exhibited more severe joint inflammation but lower levels of IL-4 compared to the control mice [110]. Although both CD4+ and CD8+ T cells are found in the inflamed synovium, CD4+ T cells make up the overwhelming part. Furthermore, depletion of CD4+ cells significantly ameliorates the course of septic arthritis in mice, whereas depletion of CD8+ T cells does not alter the course of arthritis compared to control mice [78]. Thus, it appears that CD4+ T cells are pathogenic during S. aureus septic arthritis due to their ability to produce proinflammatory cytokines such as TNF-α and IFN-γ via activated macrophages [78]. Recent results also found that CD4+ T cells promote the pathogenesis of S. aureus pneumonia and Pseudomonas aeruginosa septic arthritis [111][112].
The depletion of Tregs by anti-CD25 monoclonal antibodies aggravated the severity of septic arthritis without impact on bacterial clearance in mice [79], suggesting the protective role of Tregs in development of septic arthritis. The role of Th17 cells in S. aureus septic arthritis and sepsis is still unclear. However, IL-17 produced by Th17 cells is crucial for host defense against S. aureus skin infections [113] and septic arthritis [114].
2.6.2. Natural Killer T (NKT) Cells
NKT cells are a unique subset of T cells that can have features of both T cells and NK cells. While other subsets of T cells recognize protein antigens, NKT cells are unique in that they recognize lipids and glycolipids and make up a tiny percentage of blood T cells. Studies from S. aureus triggered sepsis indicate that NKT cells do not play any significant role in the course of the disease [115].
2.6.3. B-Cells
Unlike T-cells, B-cells do not seem to be the driving force behind the pathogenesis of S. aureus infections. Studies from murine S. aureus septic arthritis model showed that B-cell deficient mice do not differ from the wild-type controls with regards to arthritis and clearance of the bacteria, but tended to have higher mortality [80].
2.6.4. Other Cell Types
Innate lymphoid cells (ILCs) are enriched at barrier surfaces of hosts and play critical roles in maintaining tissue homeostasis and immune defense. ILCs are divided to three groups of cells with distinct immunological functions [116]. Recently, activation of type 2 ILCs (ILC2s) were shown to be protective against S. aureus sepsis in mice by promoting eosinophilia, enhancement of type 2 immunity, and consequent balance of dysregulated septic inflammatory responses [77].
Basophils are the rarest granulocyte (<1% of peripheral blood leukocytes). A very recent study demonstrated that basophil-deficient mice exhibited reduced bacterial clearance and increased mortality in a cecal ligation and puncture model of sepsis and such effect was due to reduced basophil-derived TNF production [82]. So far, the role of basophils in S. aureus septic arthritis and sepsis is still largely unknown.
Thrombocytes are the major effector cell in hemostasis. Interestingly, they also contribute to the protection against S. aureus infections through direct bacterial killing effect and by enhancing phagocytic capacity of macrophages [117]. Not surprisingly, thrombocyte depletion gave rise to reduced bacterial clearance and increased mortality in S. aureus systemic infections [81].
2.6.5. Cytokines
Several cytokines are secreted by cells of both the innate and the adaptive immunity and have different roles in S. aureus infections. Some of them will briefly be discussed below and are listed in Table 3.
Table 3. The role of cytokines in S. aureus septic arthritis and sepsis.
| Cytokine | Cell Source | Function | Role in S. aureus Infections |
|---|---|---|---|
| TNF-α | Macrophages T-cells |
Proinflammatory | Aggravate S. aureus induced septic arthritis but protective in sepsis [118]. |
| IL-1 | Macrophages Dendritic cells Endothelial cells |
Proinflammatory | Protective in S. aureus induced septic arthritis and sepsis [119]. |
| IL-6 | Macrophages and T cells | Proinflammatory | Elevated IL-6 levels in synovial fluid from septic arthritis patients [120]. The role of IL-6 was not yet assessed in animal models for septic arthritis and sepsis. |
| IL-12 | Monocytes Macrophages Dendritic cells |
Proinflammatory | Protective in S. aureus induced sepsis but not septic arthritis [121] |
| IL-4 | Th2 cells | Anti-inflammatory | Dual role in S. aureus induced septic arthritis and sepsis depending on the genetic background of the host [122][123]. |
| IL-10 | Monocytes Dendritic cells T-cells |
Anti-inflammatory | Protective in S. aureus induced septic arthritis [124]. |
| IL-17 | Th17 cells | Proinflammatory | Protective in local but not systemic S. aureus infection [114]. |
| IFN-γ | NK cells T-cells |
Proinflammatory | Protective in S. aureus induced sepsis but aggravates septic arthritis [125]. |
| Janus kinase | All cells | Proinflammatory | Protective in S. aureus septic arthritis but pathogenic in sepsis [126] |
TNF-α, one of the most studied cytokines due to its role in inflammation and many diseases, is involved in the acute-phase reaction and is mainly secreted by activated macrophages and CD4+ cells, neutrophils, mast cells, and NK cells [127].
TNF-α has a contrasting role during S. aureus infections. In patients with S. aureus arthritis, the levels of TNF-α have been shown to be highly elevated in the synovial fluid. Furthermore, it has been suggested that the levels of the cytokine could function as a predictor in determining the prognosis of the disease, with higher levels associated with worse prognosis [128]. Animal studies have shown that TNF/lymphotoxin (LT)-α double knockout mice have significantly less severe S. aureus arthritis compared to the wild-type mice [118]. Indeed, we could also show that TNFR1 knockout mice exhibited less arthritis compared to wild-type mice in antibiotic-killed S. aureus induced arthritis [73]. Anti-TNF treatment was also able to abrogate arthritis induced by antibiotic-killed S. aureus [73]. Additionally, in the S. aureus skin infection model, mice pretreated with anti-TNF agent exhibited smaller lesion (abscess) sizes compared to the control PBS-treated mice [129]. On the other hand, the lack of TNF-α was associated with impaired ability of the host to successfully clear invading S. aureus in the kidneys [110][118], thus leading to increased mortality [118].
IL-1 cytokine family is a group of eleven cytokines that play an important role in the inflammatory response. Of these, most is known regarding IL-1α IL-1β, and IL-1 receptor antagonist (IL-1Ra). IL-1α plays a central role in the induction of fever, sepsis, and inflammation and is produced by activated macrophages, neutrophils, and endothelial and epithelial cells. IL-1β is predominantly produced by activated macrophages as a proprotein and is cleaved by caspase 1 into its active mature form [130]. It plays an important role in pain, inflammation, and cartilage degradation in several inflammatory diseases [131]. In S. aureus systemic infections, IL-1R signaling is also essential to the host protection against the bacteria as shown by Hultgren et al. IL-1R−/− mice inoculated with S. aureus developed significantly higher S. aureus septic arthritis and sepsis compared to wild-type IL-1R+/+ mice [119].
IFN-γ, a potent proinflammatory cytokine, is mainly produced by NK cells and T-cells. Apart from inhibiting viral and even bacterial infections, IFN-γ activates and stimulates the macrophages to better phagocytose intracellular invaders. Different roles of IFN-γ in S. aureus triggered sepsis and septic arthritis have been described. Mice deficient of the IFN-γ receptor develop significantly more severe and frequent arthritis [132]. The mortality levels due to sepsis are also significantly increased during the early stages of the infection in the mice lacking IFN-γ receptor, whereas in later stages the reverse is true with higher mortality levels in the wild-type mice [132]. Likewise, in vivo administration of IFN-γ before and after inoculation of S. aureus improved the survival of the mice while at the same time increased the severity and frequency of arthritis [125]. The positive effects on mortality due to in vivo administration of IFN-γ correlated with improved phagocytosis and better clearance of the bacteria in both, the liver and the kidneys. On the other hand, treatment of the mice with anti-IFN-γ monoclonal antibodies attenuated the severity and frequency of arthritis due to lower levels of serum TNF-α, IL-6, and IL-1β [125].
IL-4 is an anti-inflammatory cytokine that has a role in differentiation of naïve T-cells into Th2 cells and the differentiation of B-cells into plasma cells. IL-4 promotes the cytotoxic activity of CD8+ cells, decreases the production of IFN-γ by T-cells and NK cells, and affects monocytes/macrophages by reducing their production of proinflammatory cytokines like IL-1, IL-6, and TNF-α [133]. IL-4 inhibits the intracellular killing of S. aureus in infected macrophages, without affecting phagocytosis and provides therefore a favorable milieu for survival of staphylococci [122]. In S. aureus infections, the dual role of IL-4 has been described depending on the genetic background of the host. In inbred C57BL/6 mice, IL-4 was shown to be a driving force of septic arthritis and sepsis by significantly impairing the capability of the host to clear the bacteria [122]. Enhanced staphylococcal clearance from joints and kidneys, reduced mortality, and decreased frequency of arthritis was observed in IL-4 deficient C57BL/6 mice. However, in another inbred strain, 129SV mice, the opposite was true, i.e., IL-4 protected the mice from S. aureus induced sepsis [123]. IL-4 deficient 129SV mice had a thousand times higher bacterial growth in their kidneys, significantly elevated mortality, and delayed development of septic arthritis. A differential pattern of host responsiveness was seen between these mouse strains and explanation for the discrepant outcome could lie in the variation in circulating IL-4 levels—serum IL-4 was not detectable in C57BL/6 mice whereas increased IL-4 production was observed in 129SV mice in response to S. aureus infection [123].
Although IL-6 has been shown to have some anti-inflammatory features, it is usually regarded as a proinflammatory cytokine [134]. Macrophages and T cells mainly produce IL-6 during infections or trauma. In S. aureus infections, IL-6 is usually elevated together with other proinflammatory cytokines such as IL-1β and TNF-α [128]. Synovial IL-6 together with synovial lactate and synovial fluid white blood cells count have been touted as good parameters for diagnosing septic arthritis [120].
IL-10 is an anti-inflammatory cytokine produced mainly by monocytes and to a smaller extent the lymphocytes. IL-10 promotes Th2 response while downregulating Th1 cytokine secretion by macrophages and monocytes. IL-10 plays a crucial role protecting the host against S. aureus septic arthritis by promoting bacterial clearance [124].
IL-12 is primarily produced by monocytes, macrophages, and dendritic cells. Apart from stimulating the differentiation of naive T-cells to Th1 cells, IL-12 is also involved in the production of IFN-γ and TNF-α via T-cells and NK-cells. In S. aureus infections, IL-12 is crucial for the survival of the host and deficiency of IL-12 is associated with significant accumulation of S. aureus in many organs leading ultimately to the demise of the host [121].
IL-17A is a proinflammatory cytokine produced by activated Th17 subset of T-cells. IL-17A plays a significant role in host defense against local S. aureus infections due to its ability to induce chemokines that attract and recruit neutrophils [135]. Thus, in local S. aureus infection, IL-17A-/- mice developed more synovitis and erosions and more weight loss compared to the wild-type mice [114]. On the other hand, IL-17A-/- mice did not differ from wild-type mice regarding the severity and the frequency of arthritis induced by antibiotic-killed S. aureus [73].
3. Septic Arthritis and Sepsis
Septic arthritis is a rapidly progressing and devastating joint disease caused by pathogen infection. S. aureus accounts for about 70% of the septic arthritis cases and has been shown to cause more severe infection than other microbes [136][137]. Prevalence of septic arthritis is around 6 cases per 100,000 in the general population and much higher in rheumatoid arthritis (RA) patients approaching about 70 cases per 100,000 [137]. The mortality rate is around 10–15% in non-RA patients with monoarthritis, i.e., arthritis in a single joint. Polyarthritis, on the other hand, is associated with much worse prognosis, with the mortality rate ranging up to 30–50% [137][138]. Risk factors for septic arthritis include: increasing age, pre-existing joint diseases (especially RA), intravenous drug abuse, prosthetic joints, and diabetes mellitus [137][138]. Treatment of septic arthritis consists primarily of antibiotics and joint aspiration to flush out the intra-articular pus containing both bacteria and infiltrating immune cells [139][140]. One of the devastating aspects of septic arthritis is that despite optimal antibiotic treatment, almost half of the patients will develop irreversible joint destruction [138]. Definitive diagnosis of septic arthritis requires the isolation of the microbe from the synovial fluid, although due to the fast progressing nature of the disease, physicians do not and should not wait for culture results before initiating treatment with broad spectrum antibiotics [139].
Hematogenous spread of S. aureus to the synovial membrane of joints is the most commonly reported route of acquiring septic arthritis, although the bacteria can also be introduced directly into the joints by trauma (e.g., needle accident) or spread from neighboring inflamed tissues [140]. In a retrospective study, more than 70% of septic arthritis cases were shown to be caused by hematogenous spreading [141]. The probability to develop septic arthritis following bacteremia with the optimal arthritogenic dose of S. aureus in our animal model can reach up to 80–90%. In patients, the frequency of the bone and joint infections after S. aureus bacteremia varies from 12 to 17% in different studies [142][143]. Once inside, the bacteria will employ different virulence factors to attach to the host tissue and proliferate while the host immune system will respond to the invading bacteria. It has been shown that the destruction of joints in S. aureus septic arthritis is not only caused by the invading microbes, but also by cells and molecules of the immune system, involving both the innate and adaptive immunity [140].
Sepsis is defined as the systemic inflammatory response due to an infection and is usually caused by bacteria such as S. aureus, Pseudomonas aeruginosa, and Escherichia coli [144]. S. aureus bacteremia is associated with higher mortalities than bacteremia caused by most other microbes and can develop to sepsis and severe sepsis [144]. Despite advances made in critical care and treatment, sepsis remains one of the foremost causes of death in critically ill patients. Mortality in sepsis is around 10-20% and increases significantly up to 80% if a septic shock develops [145][146].
The pathogenesis of S. aureus sepsis is multifactorial and is mediated by components of the bacteria and the exaggerated immune response mounted by the host. Bacterial superantigens can cause non-specific activation of T-cells leading to massive polyclonal T cell activation and resulting in a vast release of proinflammatory cytokines such as TNF-α and IL-1β [66][67]. PGN and LTA, cell wall components of S. aureus, can also interact with CD14 molecules through TLR2 and stimulate the release of proinflammatory cytokines (TNF-α and IL-6) and chemokines (IL-8) further potentiating the systemic inflammation in sepsis [36][147]. This is followed by a massive release of anti-inflammatory cytokines in response to the inflammation whereby the immune regulation is rendered inactive, leading to a state of immunosuppression [148]. Without a proper functioning immune system, the bacteria have free reign to proliferate and spread to different organs. Coagulation disorder, characterized by an excessive coagulation, is another attribute of sepsis. The coagulation cascade can be activated through proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 leading to disseminated intravascular coagulation (DIC) [147][149]. DIC is soon followed by thrombocytopenia, i.e., the lack of platelets in the blood resulting in massive bleeding from several sites and leading to organ failure [149]. Given together, the pathogenesis of sepsis includes systemic inflammation, loss of immune regulation, and excessive coagulation that altogether will lead to multiple organ failure, shock, and finally the demise of the host.
References
- Wertheim, H.F.; Melles, D.C.; Vos, M.C.; van Leeuwen, W.; van Belkum, A.; Verbrugh, H.A.; Nouwen, J.L. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 2005, 5, 751–762.
- Bien, J.; Sokolova, O.; Bozko, P. Characterization of Virulence Factors of Staphylococcus aureus: Novel Function of Known Virulence Factors That Are Implicated in Activation of Airway Epithelial Proinflammatory Response. J. Pathog. 2011, 2011, 601905.
- Ali, A. Biologics in Staphylococcus Aureus Arthritis. Ph.D. Thesis, University of Gothenburg, Gothenburg, Sweden, 2016.
- Nanra, J.S.; Buitrago, S.M.; Crawford, S.; Ng, J.; Fink, P.S.; Hawkins, J.; Scully, I.L.; McNeil, L.K.; Aste-Amezaga, J.M.; Cooper, D.; et al. Capsular polysaccharides are an important immune evasion mechanism for Staphylococcus aureus. Hum. Vaccin. Immunother. 2013, 9, 480–487.
- Nilsson, I.M.; Lee, J.C.; Bremell, T.; Ryden, C.; Tarkowski, A. The role of staphylococcal polysaccharide microcapsule expression in septicemia and septic arthritis. Infect. Immun. 1997, 65, 4216–4221.
- Liu, Z.Q.; Deng, G.M.; Foster, S.; Tarkowski, A. Staphylococcal peptidoglycans induce arthritis. Arthritis Res. Ther. 2001, 3, 375–380.
- Dusad, A.; Thiele, G.M.; Klassen, L.W.; Gleason, A.M.; Bauer, C.; Mikuls, T.R.; Duryee, M.J.; West, W.W.; Romberger, D.J.; Poole, J.A. Organic dust, lipopolysaccharide, and peptidoglycan inhalant exposures result in bone loss/disease. Am. J. Respir. Cell Mol. Biol. 2013, 49, 829–836.
- Nguyen, M.T.; Uebele, J.; Kumari, N.; Nakayama, H.; Peter, L.; Ticha, O.; Woischnig, A.K.; Schmaler, M.; Khanna, N.; Dohmae, N.; et al. Lipid moieties on lipoproteins of commensal and non-commensal staphylococci induce differential immune responses. Nat. Commun. 2017, 8, 2246.
- Mohammad, M.; Nguyen, M.T.; Engdahl, C.; Na, M.; Jarneborn, A.; Hu, Z.; Karlsson, A.; Pullerits, R.; Ali, A.; Gotz, F.; et al. The YIN and YANG of lipoproteins in developing and preventing infectious arthritis by Staphylococcus aureus. PLoS Pathog. 2019, 15, e1007877.
- Schmaler, M.; Jann, N.J.; Ferracin, F.; Landolt, L.Z.; Biswas, L.; Gotz, F.; Landmann, R. Lipoproteins in Staphylococcus aureus mediate inflammation by TLR2 and iron-dependent growth in vivo. J. Immunol. 2009, 182, 7110–7118.
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Hook, M. Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62.
- Hair, P.S.; Echague, C.G.; Sholl, A.M.; Watkins, J.A.; Geoghegan, J.A.; Foster, T.J.; Cunnion, K.M. Clumping factor A interaction with complement factor I increases C3b cleavage on the bacterial surface of Staphylococcus aureus and decreases complement-mediated phagocytosis. Infect. Immun. 2010, 78, 1717–1727.
- Josefsson, E.; Hartford, O.; O’Brien, L.; Patti, J.M.; Foster, T. Protection against experimental Staphylococcus aureus arthritis by vaccination with clumping factor A, a novel virulence determinant. J. Infect. Dis. 2001, 184, 1572–1580.
- Kobayashi, S.D.; DeLeo, F.R. Staphylococcus aureus protein A promotes immune suppression. MBio 2013, 4, e00764-13.
- Palmqvist, N.; Foster, T.; Tarkowski, A.; Josefsson, E. Protein A is a virulence factor in Staphylococcus aureus arthritis and septic death. Microb. Pathog. 2002, 33, 239–249.
- Palmqvist, N.; Foster, T.; Fitzgerald, J.R.; Josefsson, E.; Tarkowski, A. Fibronectin-binding proteins and fibrinogen-binding clumping factors play distinct roles in staphylococcal arthritis and systemic inflammation. J. Infect. Dis. 2005, 191, 791–798.
- Switalski, L.M.; Patti, J.M.; Butcher, W.; Gristina, A.G.; Speziale, P.; Hook, M. A collagen receptor on Staphylococcus aureus strains isolated from patients with septic arthritis mediates adhesion to cartilage. Mol. Microbiol. 1993, 7, 99–107.
- Patti, J.M.; Bremell, T.; Krajewska-Pietrasik, D.; Abdelnour, A.; Tarkowski, A.; Ryden, C.; Hook, M. The Staphylococcus aureus collagen adhesin is a virulence determinant in experimental septic arthritis. Infect. Immun. 1994, 62, 152–161.
- Vanassche, T.; Verhaegen, J.; Peetermans, W.E.; van Ryn, J.; Cheng, A.; Schneewind, O.; Hoylaerts, M.F.; Verhamme, P. Inhibition of staphylothrombin by dabigatran reduces Staphylococcus aureus virulence. J. Thromb. Haemost. 2011, 9, 2436–2446.
- Na, M.; Hu, Z.; Mohammad, M.; Stroparo, M.D.N.; Ali, A.; Fei, Y.; Jarneborn, A.; Verhamme, P.; Schneewind, O.; Missiakas, D.; et al. The Expression of von Willebrand Factor-Binding Protein Determines Joint-Invading Capacity of Staphylococcus aureus, a Core Mechanism of Septic Arthritis. mBio 2020, 11.
- Hammel, M.; Nemecek, D.; Keightley, J.A.; Thomas, G.J., Jr.; Geisbrecht, B.V. The Staphylococcus aureus extracellular adherence protein (Eap) adopts an elongated but structured conformation in solution. Protein Sci. 2007, 16, 2605–2617.
- Zecconi, A.; Scali, F. Staphylococcus aureus virulence factors in evasion from innate immune defenses in human and animal diseases. Immunol. Lett. 2013, 150, 12–22.
- Kwiecinski, J.; Jacobsson, G.; Karlsson, M.; Zhu, X.; Wang, W.; Bremell, T.; Josefsson, E.; Jin, T. Staphylokinase Promotes the Establishment of Staphylococcus aureus Skin Infections While Decreasing Disease Severity. J. Infect. Dis. 2013, 208, 990–999.
- Xu, S.X.; McCormick, J.K. Staphylococcal superantigens in colonization and disease. Front. Cell. Infect. Microbiol. 2012, 2, 52.
- McCormick, J.K.; Tripp, T.J.; Llera, A.S.; Sundberg, E.J.; Dinges, M.M.; Mariuzza, R.A.; Schlievert, P.M. Functional analysis of the TCR binding domain of toxic shock syndrome toxin-1 predicts further diversity in MHC class II/superantigen/TCR ternary complexes. J. Immunol. 2003, 171, 1385–1392.
- Abdelnour, A.; Bremell, T.; Holmdahl, R.; Tarkowski, A. Clonal expansion of T lymphocytes causes arthritis and mortality in mice infected with toxic shock syndrome toxin-1-producing staphylococci. Eur. J. Immunol. 1994, 24, 1161–1166.
- Dinges, M.M.; Orwin, P.M.; Schlievert, P.M. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 2000, 13, 16–34.
- Omoe, K.; Hu, D.L.; Ono, H.K.; Shimizu, S.; Takahashi-Omoe, H.; Nakane, A.; Uchiyama, T.; Shinagawa, K.; Imanishi, K. Emetic potentials of newly identified staphylococcal enterotoxin-like toxins. Infect. Immun. 2013, 81, 3627–3631.
- Vandenesch, F.; Lina, G.; Henry, T. Staphylococcus aureus hemolysins, bi-component leukocidins, and cytolytic peptides: A redundant arsenal of membrane-damaging virulence factors? Front. Cell. Infect. Microbiol. 2012, 2, 12.
- Nilsson, I.M.; Hartford, O.; Foster, T.; Tarkowski, A. Alpha-toxin and gamma-toxin jointly promote Staphylococcus aureus virulence in murine septic arthritis. Infect. Immun. 1999, 67, 1045–1049.
- Diep, B.A.; Chan, L.; Tattevin, P.; Kajikawa, O.; Martin, T.R.; Basuino, L.; Mai, T.T.; Marbach, H.; Braughton, K.R.; Whitney, A.R.; et al. Polymorphonuclear leukocytes mediate Staphylococcus aureus Panton-Valentine leukocidin-induced lung inflammation and injury. Proc. Natl. Acad. Sci. USA 2010, 107, 5587–5592.
- Cheung, G.Y.; Joo, H.S.; Chatterjee, S.S.; Otto, M. Phenol-soluble modulins--critical determinants of staphylococcal virulence. FEMS Microbiol. Rev. 2014, 38, 698–719.
- Periasamy, S.; Joo, H.S.; Duong, A.C.; Bach, T.H.; Tan, V.Y.; Chatterjee, S.S.; Cheung, G.Y.; Otto, M. How Staphylococcus aureus biofilms develop their characteristic structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1281–1286.
- Peschel, A.; Otto, M. Phenol-soluble modulins and staphylococcal infection. Nat. Rev. Microbiol. 2013, 11, 667–673.
- Rasigade, J.P.; Trouillet-Assant, S.; Ferry, T.; Diep, B.A.; Sapin, A.; Lhoste, Y.; Ranfaing, J.; Badiou, C.; Benito, Y.; Bes, M.; et al. PSMs of hypervirulent Staphylococcus aureus act as intracellular toxins that kill infected osteoblasts. PLoS ONE 2013, 8, e63176.
- Fournier, B.; Philpott, D.J. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 2005, 18, 521–540.
- Deng, G.M.; Nilsson, I.M.; Verdrengh, M.; Collins, L.V.; Tarkowski, A. Intra-articularly localized bacterial DNA containing CpG motifs induces arthritis. Nat. Med. 1999, 5, 702–705.
- Sparwasser, T.; Miethke, T.; Lipford, G.; Borschert, K.; Hacker, H.; Heeg, K.; Wagner, H. Bacterial DNA causes septic shock. Nature 1997, 386, 336–337.
- Watts, A.; Ke, D.; Wang, Q.; Pillay, A.; Nicholson-Weller, A.; Lee, J.C. Staphylococcus aureus strains that express serotype 5 or serotype 8 capsular polysaccharides differ in virulence. Infect. Immun. 2005, 73, 3502–3511.
- Sharif, S.; Singh, M.; Kim, S.J.; Schaefer, J. Staphylococcus aureus peptidoglycan tertiary structure from carbon-13 spin diffusion. J. Am. Chem. Soc. 2009, 131, 7023–7030.
- Vollmer, W.; Blanot, D.; de Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167.
- Willey, J.M.; Sherwood, L.; Woolverton, C.J. Prescott’s Microbiology, 9th ed.; McGraw-Hill: New York, NY, USA, 2014; p. 1.
- Grundling, A.; Schneewind, O. Cross-linked peptidoglycan mediates lysostaphin binding to the cell wall envelope of Staphylococcus aureus. J. Bacteriol. 2006, 188, 2463–2472.
- Kashyap, D.R.; Wang, M.; Liu, L.H.; Boons, G.J.; Gupta, D.; Dziarski, R. Peptidoglycan recognition proteins kill bacteria by activating protein-sensing two-component systems. Nat. Med. 2011, 17, 676–683.
- Bera, A.; Herbert, S.; Jakob, A.; Vollmer, W.; Gotz, F. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol. Microbiol. 2005, 55, 778–787.
- Sychantha, D.; Jones, C.S.; Little, D.J.; Moynihan, P.J.; Robinson, H.; Galley, N.F.; Roper, D.I.; Dowson, C.G.; Howell, P.L.; Clarke, A.J. In vitro characterization of the antivirulence target of Gram-positive pathogens, peptidoglycan O-acetyltransferase A (OatA). PLoS Pathog. 2017, 13, e1006667.
- Baranwal, G.; Mohammad, M.; Jarneborn, A.; Reddy, B.R.; Golla, A.; Chakravarty, S.; Biswas, L.; Gotz, F.; Shankarappa, S.; Jin, T.; et al. Impact of cell wall peptidoglycan O-acetylation on the pathogenesis of Staphylococcus aureus in septic arthritis. Int. J. Med. Microbiol. 2017, 307, 388–397.
- Reichmann, N.T.; Grundling, A. Location, synthesis and function of glycolipids and polyglycerolphosphate lipoteichoic acid in Gram-positive bacteria of the phylum Firmicutes. FEMS Microbiol. Lett. 2011, 319, 97–105.
- Brown, S.; Santa Maria, J.P., Jr.; Walker, S. Wall teichoic acids of gram-positive bacteria. Annu. Rev. Microbiol. 2013, 67, 313–336.
- Morath, S.; Geyer, A.; Hartung, T. Structure-function relationship of cytokine induction by lipoteichoic acid from Staphylococcus aureus. J. Exp. Med. 2001, 193, 393–397.
- Morath, S.; Stadelmaier, A.; Geyer, A.; Schmidt, R.R.; Hartung, T. Synthetic lipoteichoic acid from Staphylococcus aureus is a potent stimulus of cytokine release. J. Exp. Med. 2002, 195, 1635–1640.
- Nguyen, M.T.; Gotz, F. Lipoproteins of Gram-Positive Bacteria: Key Players in the Immune Response and Virulence. Microbiol. Mol. Biol. Rev. 2016, 80, 891–903.
- Sheldon, J.R.; Heinrichs, D.E. The iron-regulated staphylococcal lipoproteins. Front. Cell. Infect. Microbiol. 2012, 2, 41.
- Hammer, N.D.; Skaar, E.P. Molecular mechanisms of Staphylococcus aureus iron acquisition. Annu. Rev. Microbiol. 2011, 65, 129–147.
- Mohammad, M.; Hu, Z.; Ali, A.; Kopparapu, P.K.; Na, M.; Jarneborn, A.; Stroparo, M.d.N.; Nguyen, M.-T.; Karlsson, A.; Götz, F.; et al. The role of Staphylococcus aureus lipoproteins in hematogenous septic arthritis. Sci. Rep. 2020, 10, 7936.
- Nguyen, M.T.; Kraft, B.; Yu, W.; Demircioglu, D.D.; Hertlein, T.; Burian, M.; Schmaler, M.; Boller, K.; Bekeredjian-Ding, I.; Ohlsen, K.; et al. The νSaα Specific Lipoprotein Like Cluster (lpl) of S. aureus USA300 Contributes to Immune Stimulation and Invasion in Human Cells. PLoS Pathog. 2015, 11, e1004984.
- Joh, D.; Wann, E.R.; Kreikemeyer, B.; Speziale, P.; Hook, M. Role of fibronectin-binding MSCRAMMs in bacterial adherence and entry into mammalian cells. Matrix Biol. 1999, 18, 211–223.
- An, Y.H.; Friedman, R.J. Handbook of Bacterial Adhesion Principles, Methods, and Applications; Humana Press: Totowa, NJ, USA, 2000; Volume xvi, 645p.
- Gomez, M.I.; O’Seaghdha, M.; Magargee, M.; Foster, T.J.; Prince, A.S. Staphylococcus aureus protein A activates TNFR1 signaling through conserved IgG binding domains. J. Biol. Chem. 2006, 281, 20190–20196.
- Chavakis, T.; Wiechmann, K.; Preissner, K.T.; Herrmann, M. Staphylococcus aureus interactions with the endothelium: The role of bacterial “secretable expanded repertoire adhesive molecules” (SERAM) in disturbing host defense systems. Thromb. Haemost. 2005, 94, 278–285.
- Claes, J.; Vanassche, T.; Peetermans, M.; Liesenborghs, L.; Vandenbriele, C.; Vanhoorelbeke, K.; Missiakas, D.; Schneewind, O.; Hoylaerts, M.F.; Heying, R.; et al. Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von Willebrand factor-binding protein. Blood 2014, 124, 1669–1676.
- Claes, J.; Liesenborghs, L.; Peetermans, M.; Veloso, T.R.; Missiakas, D.; Schneewind, O.; Mancini, S.; Entenza, J.M.; Hoylaerts, M.F.; Heying, R.; et al. Clumping factor A, von Willebrand factor-binding protein and von Willebrand factor anchor Staphylococcus aureus to the vessel wall. J. Thromb. Haemost. 2017, 15, 1009–1019.
- Bardoel, B.W.; Vos, R.; Bouman, T.; Aerts, P.C.; Bestebroer, J.; Huizinga, E.G.; Brondijk, T.H.; van Strijp, J.A.; de Haas, C.J. Evasion of Toll-like receptor 2 activation by staphylococcal superantigen-like protein 3. J. Mol. Med. (Berl.) 2012, 90, 1109–1120.
- Ibberson, C.B.; Jones, C.L.; Singh, S.; Wise, M.C.; Hart, M.E.; Zurawski, D.V.; Horswill, A.R. Staphylococcus aureus hyaluronidase is a CodY-regulated virulence factor. Infect. Immun. 2014, 82, 4253–4264.
- Kwiecinski, J.; Peetermans, M.; Liesenborghs, L.; Na, M.; Bjornsdottir, H.; Zhu, X.; Jacobsson, G.; Johansson, B.R.; Geoghegan, J.A.; Foster, T.J.; et al. Staphylokinase Control of Staphylococcus aureus Biofilm Formation and Detachment Through Host Plasminogen Activation. J. Infect. Dis. 2016, 213, 139–148.
- Fraser, J.D. Clarifying the mechanism of superantigen toxicity. PLoS Biol. 2011, 9, e1001145.
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: An update. Annu. Rev. Microbiol. 2001, 55, 77–104.
- Marrack, P.; Blackman, M.; Kushnir, E.; Kappler, J. The toxicity of staphylococcal enterotoxin B in mice is mediated by T cells. J. Exp. Med. 1990, 171, 455–464.
- Kaempfer, R.; Arad, G.; Levy, R.; Hillman, D.; Nasie, I.; Rotfogel, Z. CD28: Direct and critical receptor for superantigen toxins. Toxins (Basel) 2013, 5, 1531–1542.
- Gouaux, E. alpha-Hemolysin from Staphylococcus aureus: An archetype of beta-barrel, channel-forming toxins. J. Struct. Biol. 1998, 121, 110–122.
- Krieg, A.M.; Yi, A.K.; Matson, S.; Waldschmidt, T.J.; Bishop, G.A.; Teasdale, R.; Koretzky, G.A.; Klinman, D.M. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995, 374, 546–549.
- Deng, G.M.; Liu, Z.Q.; Tarkowski, A. Intracisternally localized bacterial DNA containing CpG motifs induces meningitis. J. Immunol. 2001, 167, 4616–4626.
- Ali, A.; Zhu, X.; Kwiecinski, J.; Gjertsson, I.; Lindholm, C.; Iwakura, Y.; Wang, X.; Lycke, N.; Josefsson, E.; Pullerits, R.; et al. Antibiotic-killed Staphylococcus aureus induces destructive arthritis in mice. Arthritis Rheumatol. 2015, 67, 107–116.
- Verdrengh, M.; Tarkowski, A. Role of neutrophils in experimental septicemia and septic arthritis induced by Staphylococcus aureus. Infect. Immun. 1997, 65, 2517–2521.
- Verdrengh, M.; Tarkowski, A. Role of macrophages in Staphylococcus aureus-induced arthritis and sepsis. Arthritis Rheum. 2000, 43, 2276–2282.
- Nilsson, N.; Bremell, T.; Tarkowski, A.; Carlsten, H. Protective role of NK1.1+ cells in experimental Staphylococcus aureus arthritis. Clin. Exp. Immunol. 1999, 117, 63–69.
- Krishack, P.A.; Louviere, T.J.; Decker, T.S.; Kuzel, T.G.; Greenberg, J.A.; Camacho, D.F.; Hrusch, C.L.; Sperling, A.I.; Verhoef, P.A. Protection against Staphylococcus aureus bacteremia-induced mortality depends on ILC2s and eosinophils. JCI Insight 2019, 4.
- Abdelnour, A.; Bremell, T.; Holmdahl, R.; Tarkowski, A. Role of T lymphocytes in experimental Staphylococcus aureus arthritis. Scand. J. Immunol. 1994, 39, 403–408.
- Bergmann, B.; Fei, Y.; Jirholt, P.; Hu, Z.; Bergquist, M.; Ali, A.; Lindholm, C.; Ekwall, O.; Churlaud, G.; Klatzmann, D.; et al. Pre-treatment with IL2 gene therapy alleviates Staphylococcus aureus arthritis in mice. BMC Infect. Dis. 2020, 20, 185.
- Gjertsson, I.; Hultgren, O.H.; Stenson, M.; Holmdahl, R.; Tarkowski, A. Are B lymphocytes of importance in severe Staphylococcus aureus infections? Infect. Immun. 2000, 68, 2431–2434.
- Wuescher, L.M.; Takashima, A.; Worth, R.G. A novel conditional platelet depletion mouse model reveals the importance of platelets in protection against Staphylococcus aureus bacteremia. J. Thromb. Haemost. 2015, 13, 303–313.
- Piliponsky, A.M.; Shubin, N.J.; Lahiri, A.K.; Truong, P.; Clauson, M.; Niino, K.; Tsuha, A.L.; Nedospasov, S.A.; Karasuyama, H.; Reber, L.L.; et al. Basophil-derived tumor necrosis factor can enhance survival in a sepsis model in mice. Nat. Immunol. 2019, 20, 129–140.
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801.
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175.
- Abbas, A.K.; Lichtman, A.H. Basic Immunology: Functions and Disorders of the Immune System, 3rd ed.; Saunders/Elsevier: Philadelphia, PA, USA, 2009.
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393.
- Molne, L.; Verdrengh, M.; Tarkowski, A. Role of neutrophil leukocytes in cutaneous infection caused by Staphylococcus aureus. Infect. Immun. 2000, 68, 6162–6167.
- Mills, C.D. Anatomy of a discovery: m1 and m2 macrophages. Front. Immunol. 2015, 6, 212.
- Rath, M.; Muller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532.
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969.
- Makita, N.; Hizukuri, Y.; Yamashiro, K.; Murakawa, M.; Hayashi, Y. IL-10 enhances the phenotype of M2 macrophages induced by IL-4 and confers the ability to increase eosinophil migration. Int. Immunol. 2015, 27, 131–141.
- Dey, A.; Allen, J.; Hankey-Giblin, P.A. Ontogeny and polarization of macrophages in inflammation: Blood monocytes versus tissue macrophages. Front. Immunol. 2014, 5, 683.
- Tanaka, Y.; Nakayamada, S.; Okada, Y. Osteoblasts and osteoclasts in bone remodeling and inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 325–328.
- Ogata, Y.; Kukita, A.; Kukita, T.; Komine, M.; Miyahara, A.; Miyazaki, S.; Kohashi, O. A novel role of IL-15 in the development of osteoclasts: Inability to replace its activity with IL-2. J. Immunol. 1999, 162, 2754–2760.
- Djaafar, S.; Pierroz, D.D.; Chicheportiche, R.; Zheng, X.X.; Ferrari, S.L.; Ferrari-Lacraz, S. Inhibition of T cell-dependent and RANKL-dependent osteoclastogenic processes associated with high levels of bone mass in interleukin-15 receptor-deficient mice. Arthritis Rheum. 2010, 62, 3300–3310.
- Henningsson, L.; Jirholt, P.; Bogestal, Y.R.; Eneljung, T.; Adiels, M.; Lindholm, C.; McInnes, I.; Bulfone-Paus, S.; Lerner, U.H.; Gjertsson, I. Interleukin 15 mediates joint destruction in Staphylococcus aureus arthritis. J. Infect. Dis. 2012, 206, 687–696.
- Verdrengh, M.; Bokarewa, M.; Ohlsson, C.; Stolina, M.; Tarkowski, A. RANKL-targeted therapy inhibits bone resorption in experimental Staphylococcus aureus-induced arthritis. Bone 2010, 46, 752–758.
- Deng, G.M.; Verdrengh, M.; Liu, Z.Q.; Tarkowski, A. The major role of macrophages and their product tumor necrosis factor alpha in the induction of arthritis triggered by bacterial DNA containing CpG motifs. Arthritis Rheum. 2000, 43, 2283–2289.
- Small, C.L.; McCormick, S.; Gill, N.; Kugathasan, K.; Santosuosso, M.; Donaldson, N.; Heinrichs, D.E.; Ashkar, A.; Xing, Z. NK cells play a critical protective role in host defense against acute extracellular Staphylococcus aureus bacterial infection in the lung. J. Immunol. 2008, 180, 5558–5568.
- Laarman, A.J.; Ruyken, M.; Malone, C.L.; van Strijp, J.A.; Horswill, A.R.; Rooijakkers, S.H. Staphylococcus aureus metalloprotease aureolysin cleaves complement C3 to mediate immune evasion. J. Immunol. 2011, 186, 6445–6453.
- Laarman, A.; Milder, F.; van Strijp, J.; Rooijakkers, S. Complement inhibition by gram-positive pathogens: Molecular mechanisms and therapeutic implications. J. Mol. Med. (Berl.) 2010, 88, 115–120.
- Sakiniene, E.; Bremell, T.; Tarkowski, A. Complement depletion aggravates Staphylococcus aureus septicaemia and septic arthritis. Clin. Exp. Immunol. 1999, 115, 95–102.
- Na, M.; Jarneborn, A.; Ali, A.; Welin, A.; Magnusson, M.; Stokowska, A.; Pekna, M.; Jin, T. Deficiency of the complement component 3 but not factor B aggravates Staphylococcus aureus septic arthritis in mice. Infect. Immun. 2016.
- Andersen, M.H.; Schrama, D.; Thor Straten, P.; Becker, J.C. Cytotoxic T cells. J. Investig. Dermatol. 2006, 126, 32–41.
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787.
- Tesmer, L.A.; Lundy, S.K.; Sarkar, S.; Fox, D.A. Th17 cells in human disease. Immunol. Rev. 2008, 223, 87–113.
- Kidd, P. Th1/Th2 balance: The hypothesis, its limitations, and implications for health and disease. Altern. Med. Rev. 2003, 8, 223–246.
- Romagnani, S. Th1/Th2 cells. Inflamm. Bowel Dis. 1999, 5, 285–294.
- Oosterwegel, M.A.; Mandelbrot, D.A.; Boyd, S.D.; Lorsbach, R.B.; Jarrett, D.Y.; Abbas, A.K.; Sharpe, A.H. The role of CTLA-4 in regulating Th2 differentiation. J. Immunol. 1999, 163, 2634–2639.
- Ali, A.; Welin, A.; Schwarze, J.C.; Svensson, M.N.; Na, M.; Jarneborn, A.; Magnusson, M.; Mohammad, M.; Kwiecinski, J.; Josefsson, E.; et al. CTLA4 Immunoglobulin but Not Anti-Tumor Necrosis Factor Therapy Promotes Staphylococcal Septic Arthritis in Mice. J. Infect. Dis. 2015, 212, 1308–1316.
- Parker, D.; Ryan, C.L.; Alonzo, F., 3rd; Torres, V.J.; Planet, P.J.; Prince, A.S. CD4+ T cells promote the pathogenesis of Staphylococcus aureus pneumonia. J. Infect. Dis. 2015, 211, 835–845.
- Jin, T.; Mohammad, M.; Hu, Z.; Fei, Y.; Moore, E.R.B.; Pullerits, R.; Ali, A. A novel mouse model for septic arthritis induced by Pseudomonas aeruginosa. Sci. Rep. 2019, 9, 16868.
- Cho, J.S.; Pietras, E.M.; Garcia, N.C.; Ramos, R.I.; Farzam, D.M.; Monroe, H.R.; Magorien, J.E.; Blauvelt, A.; Kolls, J.K.; Cheung, A.L.; et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin. Investig. 2010, 120, 1762–1773.
- Henningsson, L.; Jirholt, P.; Lindholm, C.; Eneljung, T.; Silverpil, E.; Iwakura, Y.; Linden, A.; Gjertsson, I. Interleukin-17A during local and systemic Staphylococcus aureus-induced arthritis in mice. Infect. Immun. 2010, 78, 3783–3790.
- Kwiecinski, J.; Rhost, S.; Lofbom, L.; Blomqvist, M.; Mansson, J.E.; Cardell, S.L.; Jin, T. Sulfatide attenuates experimental Staphylococcus aureus sepsis through a CD1d-dependent pathway. Infect. Immun. 2013, 81, 1114–1120.
- Sonnenberg, G.F.; Hepworth, M.R. Functional interactions between innate lymphoid cells and adaptive immunity. Nat. Rev. Immunol. 2019, 19, 599–613.
- Ali, R.A.; Wuescher, L.M.; Dona, K.R.; Worth, R.G. Platelets Mediate Host Defense against Staphylococcus aureus through Direct Bactericidal Activity and by Enhancing Macrophage Activities. J. Immunol. 2017, 198, 344–351.
- Hultgren, O.; Eugster, H.P.; Sedgwick, J.D.; Korner, H.; Tarkowski, A. TNF/lymphotoxin-alpha double-mutant mice resist septic arthritis but display increased mortality in response to Staphylococcus aureus. J. Immunol. 1998, 161, 5937–5942.
- Hultgren, O.H.; Svensson, L.; Tarkowski, A. Critical role of signaling through IL-1 receptor for development of arthritis and sepsis during Staphylococcus aureus infection. J. Immunol. 2002, 168, 5207–5212.
- Lenski, M.; Scherer, M.A. The significance of interleukin-6 and lactate in the synovial fluid for diagnosing native septic arthritis. Acta Orthop. Belg. 2014, 80, 18–25.
- Hultgren, O.H.; Stenson, M.; Tarkowski, A. Role of IL-12 in Staphylococcus aureus-triggered arthritis and sepsis. Arthritis Res. 2001, 3, 41–47.
- Hultgren, O.; Kopf, M.; Tarkowski, A. Staphylococcus aureus-induced septic arthritis and septic death is decreased in IL-4-deficient mice: Role of IL-4 as promoter for bacterial growth. J. Immunol. 1998, 160, 5082–5087.
- Hultgren, O.; Kopf, M.; Tarkowski, A. Outcome of Staphylococcus aureus-triggered sepsis and arthritis in IL-4-deficient mice depends on the genetic background of the host. Eur. J. Immunol. 1999, 29, 2400–2405.
- Gjertsson, I.; Hultgren, O.H.; Tarkowski, A. Interleukin-10 ameliorates the outcome of Staphylococcus aureus arthritis by promoting bacterial clearance. Clin. Exp. Immunol. 2002, 130, 409–414.
- Zhao, Y.X.; Nilsson, I.M.; Tarkowski, A. The dual role of interferon-gamma in experimental Staphylococcus aureus septicaemia versus arthritis. Immunology 1998, 93, 80–85.
- Jarneborn, A.; Mohammad, M.; Engdahl, C.; Hu, Z.; Na, M.; Ali, A.; Jin, T. Tofacitinib treatment aggravates Staphylococcus aureus septic arthritis, but attenuates sepsis and enterotoxin induced shock in mice. Sci. Rep. 2020, 10, 10891.
- Davis, L.S.; Hutcheson, J.; Mohan, C. The role of cytokines in the pathogenesis and treatment of systemic lupus erythematosus. J. Interferon Cytokine Res. 2011, 31, 781–789.
- Osiri, M.; Ruxrungtham, K.; Nookhai, S.; Ohmoto, Y.; Deesomchok, U. IL-1beta, IL-6 and TNF-alpha in synovial fluid of patients with non-gonococcal septic arthritis. Asian Pac. J. Allergy Immunol. 1998, 16, 155–160.
- Na, M.; Wang, W.; Fei, Y.; Josefsson, E.; Ali, A.; Jin, T. Both anti-TNF and CTLA4 Ig treatments attenuate the disease severity of staphylococcal dermatitis in mice. PLoS ONE 2017, 12, e0173492.
- Jacques, C.; Gosset, M.; Berenbaum, F.; Gabay, C. The role of IL-1 and IL-1Ra in joint inflammation and cartilage degradation. Vitam. Horm. 2006, 74, 371–403.
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732.
- Zhao, Y.X.; Tarkowski, A. Impact of interferon-gamma receptor deficiency on experimental Staphylococcus aureus septicemia and arthritis. J. Immunol. 1995, 155, 5736–5742.
- O’Garra, A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity 1998, 8, 275–283.
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 2011, 1813, 878–888.
- Ivanov, S.; Linden, A. Interleukin-17 as a drug target in human disease. Trends Pharmacol. Sci. 2009, 30, 95–103.
- Sharff, K.A.; Richards, E.P.; Townes, J.M. Clinical management of septic arthritis. Curr. Rheumatol. Rep. 2013, 15, 332.
- Tarkowski, A. Infection and musculoskeletal conditions: Infectious arthritis. Best Pract. Res. Clin. Rheumatol. 2006, 20, 1029–1044.
- Goldenberg, D.L. Septic arthritis. Lancet 1998, 351, 197–202.
- Garcia-Arias, M.; Balsa, A.; Mola, E.M. Septic arthritis. Best Pract. Res. Clin. Rheumatol. 2011, 25, 407–421.
- Mathews, C.J.; Weston, V.C.; Jones, A.; Field, M.; Coakley, G. Bacterial septic arthritis in adults. Lancet 2010, 375, 846–855.
- Morgan, D.S.; Fisher, D.; Merianos, A.; Currie, B.J. An 18 year clinical review of septic arthritis from tropical Australia. Epidemiol. Infect. 1996, 117, 423–428.
- Fowler, V.G., Jr.; Sanders, L.L.; Sexton, D.J.; Kong, L.; Marr, K.A.; Gopal, A.K.; Gottlieb, G.; McClelland, R.S.; Corey, G.R. Outcome of Staphylococcus aureus bacteremia according to compliance with recommendations of infectious diseases specialists: Experience with 244 patients. Clin. Infect. Dis. 1998, 27, 478–486.
- Marr, K.A.; Kong, L.; Fowler, V.G.; Gopal, A.; Sexton, D.J.; Conlon, P.J.; Corey, G.R. Incidence and outcome of Staphylococcus aureus bacteremia in hemodialysis patients. Kidney Int. 1998, 54, 1684–1689.
- Naber, C.K. Staphylococcus aureus bacteremia: Epidemiology, pathophysiology, and management strategies. Clin. Infect. Dis. 2009, 48 (Suppl. 4), S231–S237.
- Holland, T.L.; Arnold, C.; Fowler, V.G., Jr. Clinical management of Staphylococcus aureus bacteremia: A review. JAMA 2014, 312, 1330–1341.
- Martin, G.S. Sepsis, severe sepsis and septic shock: Changes in incidence, pathogens and outcomes. Expert. Rev. Anti Infect. Ther. 2012, 10, 701–706.
- Van Amersfoort, E.S.; Van Berkel, T.J.; Kuiper, J. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clin. Microbiol. Rev. 2003, 16, 379–414.
- Huttunen, R.; Aittoniemi, J. New concepts in the pathogenesis, diagnosis and treatment of bacteremia and sepsis. J. Infect. 2011, 63, 407–419.
- Semeraro, N.; Ammollo, C.T.; Semeraro, F.; Colucci, M. Sepsis-associated disseminated intravascular coagulation and thromboembolic disease. Mediterr. J. Hematol. Infect. Dis. 2010, 2, e2010024.

