 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Joshua Choe | + 2342 word(s) | 2342 | 2021-03-29 10:10:55 | | | |
| 2 | Lily Guo | Meta information modification | 2342 | 2021-04-06 03:09:03 | | |
Video Upload Options
Squamous cell carcinomas (SCCs) are cancers that arise from both stratified squamous and non-squamous epithelium of diverse anatomical sites and collectively represent one of the most frequent solid tumors. Despite such heterogeneous anatomical origins, SCCs have unified oncogenic and metabolic features centered around maintaining redox homeostasis that may ultimately be attractive therapeutic targets.
1. Introduction
Squamous epithelia are comprised of proliferative basal layers and increasingly differentiated suprabasal layers in the outermost layer of the skin, inner mouth, esophagus, and anogenital regions and may also develop from squamous metaplasia, a replacement of non-squamous epithelium by squamous epithelium, induced by injury or other stress conditions in the respiratory and urinary tracts [1][2]. SCCs can arise from both stratified squamous epithelium and non-squamous epithelium across many anatomical sites including most frequently in the skin, lung, head and neck, esophagus, and cervix [2]. In addition, SCCs may develop from non-squamous epithelial tissue through squamous transdifferentiation [2][3][4][5]. Despite the diverse locations in which they are found, SCCs commonly originate from proliferative basal cells. In contrast, other cell types of squamous epithelia, such as transit-amplifying progenitors and terminally differentiated, non-proliferative epithelial cells, are unable to generate malignant tumors even with the introduction of oncogenic mutations [6][7][8].
Recent comprehensive genomic characterizations of common SCCs demonstrate similar mutational landscapes even across different SCCs including aberrations in TP53, TP63, SOX2, PIK3CA, KEAP1, and NFE2L2 genes [9][10][11][12][13][14][15]. However, targeted therapies remain limited, and approved RTK inhibitors such as EGFR inhibitor cetuximab only modestly improve survival in SCC patients and inevitably lead to clinical resistance [2][3][4][5][16][17][18]. Although with current standards of treatment including surgical resection and radiation of low-risk or early stage SCCs are highly curable (~95–98%), locally advanced and metastatic SCCs retain poor five-year survival rates. These advanced cancers remain difficult to treat, and only highly toxic chemotherapy regimens, such as 5-fluorouracil and platinum-based agents, are available for these cases. As a result of high cellular heterogeneity and their intrinsic ability to enter into drug-resistant TGF-beta-mediated quiescent phases, SCCs are also difficult to treat with monotherapeutic approaches and frequently display chemoresistance [17][19]. A preponderance of recent evidence characterizing an SCC-specific convergence of oncogenic signaling and metabolism may suggest a novel paradigm in the treatment of advanced SCC: synergistically targeting the nexus of oncogenic and metabolic pathways [16][18][20].
Reactive oxygen species (ROS) come in many flavors, among which superoxide, hydrogen peroxide, and hydroxyl radicals are most well studied in the context of cancer, and can be produced from both endogenous and exogenous sources [21]. ROS play a complex and as yet incompletely understood role in cancer, playing both tumor promoting and tumor suppressive roles depending on ROS levels and stage of tumor progression [22]. At moderate levels, ROS may promote tumor formation, increased proliferation and survival signaling, genomic instability, and increased motility. Excessive ROS, however, induces arrest, senescence, and death in cancers. Specifically, some types of ROS can drive DNA damage in normal tissues, which can result in the first mutations that can set a cell on the path to tumor formation [23]. Further, ROS can activate pro-cancer signaling pathways including p38 mediated MAP kinase activation and hypoxia inducible factor (HIF) stabilization [24][25][26]. Conversely, when left unchecked, ROS levels can cause cell death through a variety of mechanisms that may be exploited to treat cancer [27]. However, tumor metabolic reprogramming can help to protect cancers from ROS toxicity by driving the production of the cell’s primary antioxidant glutathione (GSH), and the NADPH required to recycle oxidized GSH back to its reduced form [28]. Recently, analysis into the composition of cutaneous SCCs (CSCC) revealed distinct subpopulations within tumors that recapitulate normal keratinocyte populations, basal, cycling, and differentiated, but have acquired novel oncogenic characteristics including the upregulation of glycolysis and response to ROS and a downregulation of apoptosis [29]. Although the specific contributions of various ROS species and their regulation in different cellular compartments are undoubtedly relevant in cancer, here we focus on ROS as damaging agents that necessitate cellular adaptation in SCCs.
2. Commonalities in Squamous Etiology and Mechanisms of Carcinogenesis
Epidemiological studies have long demonstrated a strong association between SCCs and a myriad of environmental risk factors including UV exposure, smoking, alcohol consumption, and human papilloma virus (HPV) infection, among others (Table 1) [30][31][32]. Despite incredible diversity in risk factors causally associated with SCCs, key mechanistic similarities in major risk factors driving squamous-specific tumorigenesis exist even across SCCs of different anatomical origin [2]. Specifically, induction of ROS is a feature common amongst the myriad SCC risk factors. Thus, oxidative stress may be one of the key contributors in the oncogenesis of SCCs in conjunction with other mutagenic events that ultimately drive the acquisition of antioxidant defense strategies.
Table 1. Summary of epidemiological risk factors for SCCs, mechanism of action, and common genetic aberration associated with such.
| Risk Factor | SCC Subtype | Mechanism of Action | Common Gene Mutations |
|---|---|---|---|
| Alcohol Consumption |
HN, Esophageal | -Acetaldehyde-DNA adduct formation -Oxidative stress induced DNA damage -Lipid peroxidation products -Inhibition of DNA repair |
TP53, NOTCH1, CDKN2A, CCND1, PIK3CA, TAF1L |
| Cigarette Smoking | HN, Lung, Esophageal | -Oxidative stress induced DNA damage -Mutations in guanine base pairs resulting in G/T substitutions |
TP53, TP63, EGFR, PIK3CA |
| UV exposure, UV radiation (UVA, UVB), |
Skin (cutaneous) | -DNA damage resulting in C/T substitutions -Production of reactive oxygen species (ROS). 8-oxo-dG formation; G/T transversions form during DNA replication. |
TP53, NOTCH1, 2 CDKN2A, FAT1, CASP8, HRAS, AJUBA, KMT2D |
| Infections EBV |
HN | -Viral nuclear proteins EBNA2, EBNA3A, 3B, 3C activate MAPK/Jun, PI3K/AKT and B-catenin-signaling pathways |
TP53, CDKN2A, CCND1 |
| HPV | HN, Cervical | -E6 and E7 oncoproteins target p53 and pRb tumor suppressor functions |
TP53, PIK3CA, FBXW7, NOTCH2, RB |
3. Metabolic Dependencies in Squamous Cell Carcinomas
3.1. Glucose
Tumors engage in metabolic reprogramming to fulfill cellular bioenergetic and anabolic needs as well as to fuel antioxidant production and generate redox cofactors. Emerging evidence has identified distinct metabolic phenotypes characteristic of SCCs. Diverting glycolytic flux into antioxidant-generating pathways, such as the pentose phosphate pathway (PPP) and de novo serine biosynthesis, remain critical strategies cancers utilize to combat toxic oxidative stress and may thereby metabolically reprogram to upregulate glycolysis and glucose influx [33]. Goodwin et al. demonstrate that LSCCs are uniquely reliant on glucose uptake mediated by significantly elevated glucose transporter 1 (GLUT1) with high GLUT1-expressing SCC patients exhibiting decreased survival [34]. This addiction to glucose renders LSCCs exquisitely vulnerable to glycolytic inhibition in vitro and in vivo while glucose remains dispensable for lung ADCs. Furthermore, multiple studies have identified higher F18-fluorodeoxyglucose (18F-FDG) uptake via 18F-FDG PET imaging in patients with LSCC tumors as compared to ADC tumors as well as higher risk of risk of recurrence and lower survival in tumors with high uptake of 18F-FDG [34][35][36][37][38].
This distinct metabolic feature, rather than a characteristic isolated to LSCCs specifically, is a convergent phenotype intrinsically associated with SCCs as a whole. Hsieh et al. reveal that head and neck, lung, esophageal, and cervical SCCs are the highest GLUT1-expressing cancers and critically rely on GLUT1-mediated elevated glucose influx to fuel antioxidant production, ultimately indicating a squamous lineage-specific convergent phenotype [39]. The essential contribution of GLUT1 overexpression to antioxidant production is demonstrated by the clear association of GLUT1 overexpression with resistance to radiation therapy, which primarily exerts cytotoxic effects via direct genomic damage and indirect free radical generation, in oral, cervical, and head and neck SCCs [40][41][42]. By redirecting enhanced glucose flux driven by GLUT1 overexpression into the PPP, a major cellular source of the NADPH used to recycle oxidized GSH, and the de novo serine biosynthesis pathway, which can provide the glycine component of glutathione and mitochondrial NADPH, SCCs generate elevated NADPH and GSH to ultimately maintain redox homeostasis and heighten antioxidant defenses (Figure 1) [39][43]. Inhibiting GSH metabolism via buthionine sulfoxamine treatment attenuated ESCC progression in vivo and enhanced cell killing in conjunction with thioredoxin inhibition in HNSCC cell lines [44][45]. Inhibition of the PPP via glucose-6-phosphate dehydrogenase (G6PD) blockade induced cell death in HNSCC cell lines by precipitating ER stress and accumulation of ROS [46]. Moreover, integrative transcriptomic and metabolomic analyses in vivo of lung ADC and SCC tumors reveal a distinctive metabolic signature of SCCs characterized by enhanced glucose catabolism into glycolysis, Krebs cycle, and pentose phosphate, GSH, and serine biosynthesis [47].
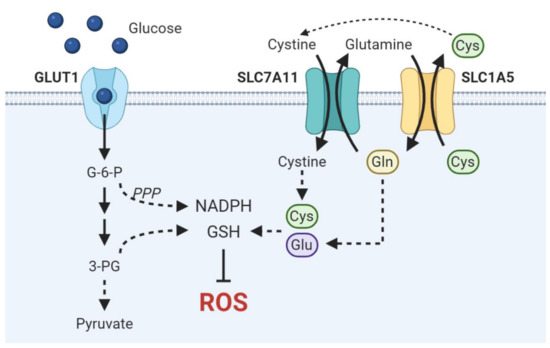
Figure 1. Schematic overview of metabolic pathways critical in driving antioxidant production in SCCs. Enhanced glucose uptake fuels NADPH and GSH synthesis via the pentose phosphate pathway and de novo serine biosynthesis, respectively. Cysteine and glutamate are necessary to synthesize the GSH precursor γ-glutamylcysteine. SLC7A11 and SLC1A5 are functionally coupled to drive cystine import.
HPV-positive HNSCCs may be the exception to this metabolic commonality. HPV oncoproteins E6 and E7 have traditionally been thought to increase glucose uptake and glycolysis by inhibiting p53, interacting with c-Myc, binding PKM2, and enhancing both HIF1 and HK2 expression, among other mechanisms [48][49][50][51]. However, recent evidence demonstrates that HPV-positive HNSCCs exhibit comparatively decreased glycolysis and lower expression levels of genes involved in glycolysis [52][53][54]. Although the conflicting functions of HPV in carbohydrate metabolism warrants further investigation, emerging evidence suggests that HPV-associated cancers may need to be considered as metabolically distinct from their counterparts.
3.2. Glutamine
While redox homeostasis is accomplished primarily through glycolytic upregulation in SCC-specific metabolic programs, SCCs may also exploit amino acid transport to drive antioxidant anabolism and impart metabolic flexibility under glucose limiting conditions. Flores et al. suggest that lactate dehydrogenase A (LDHA) and aerobic glycolysis may be dispensable in CSCCs as LDHA abrogation demonstrates no effect on tumor initiation or progression despite reduced glucose uptake and glycolytic intermediates [55]. However, they further demonstrate that LDHA-deficient tumors exhibited increased compensatory glutamine uptake, glutaminase activity, and reductive glutamine metabolism. This result demonstrates the flexibility by which SCCs maintain redox homeostasis and implicates the importance of maintaining intracellular antioxidant pools in SCCs. Glutamate and cysteine are essential precursors in de novo glutathione synthesis. ASCT2 (SLC1A5) mediates the influx of glutamine in exchange for cysteine efflux while cystine-glutamate antiporter xCT (SLC7A11) couples cystine import with glutamate export. Tumors may functionally couple these amino acid transporters to balance both intracellular cysteine and glutamate necessary for GSH synthesis (Figure 1) [56][57]. Suggestive of this molecular dependence is the observation that HNSCC and ESCCs are addicted to glutamine, the uptake of which is mediated by upregulated ASCT2 expression in HNSCC and necessary for GSH synthesis [58][59]. Furthermore, high ASCT2 expression is associated with decreased survival in both OSCC and HNSCC patients [59][60]. Work by Momcilovic et al. demonstrate that suppression of glycolysis by chronic mTOR inhibition elicits an adaptive upregulation of glutaminolysis via the GSK3 signaling axis in LSCC and HNSCCs, thereby not only defining a unifying metabolic signature of SCCs but also highlighting the metabolic plasticity by which SCCs sustain antioxidant anabolism [61].
3.3. Lactate and the Tumor Microenvironment
The metabolic reprogramming of the tumor microenvironment and the associated metabolic crosstalk between stroma and cancer cells also contributes to antioxidant defenses and tumor aggressiveness. Recent evidence demonstrates the cigarette smoke-induced metabolic reprogramming of cancer-associated fibroblasts (CAFs) towards a highly glycolytic phenotype. The highly glycolytic phenotype induces metabolic coupling and enhances the aggressiveness in HNSCCs, implicating oxidative stress in not only altering squamous metabolism but also shifting tumor stroma into a pro-oncogenic metabolic environment [62]. Additionally, CAFs may facilitate the glycolytic switch of HNSCCs by secreting HGF, which in turn induces bFGF secretion from HNSCCs to promote fibroblast proliferation [63]. Paracrine signaling and metabolic crosstalk within the tumor microenvironment results in concomitant increases in glycolysis in both tumor-associated stroma and tumor cells, thereby acidifying the extracellular space via monocarboxylate transporter-mediated lactate excretion [64]. This acidic milieu promotes immune evasion and drives tumor invasion especially at the tumor-stroma interface by increasing matrix metalloprotease (MMP) expression and rewiring of the transcriptome involved in the expression of an alternative splicing-dependent tumor invasion program [65][66]. Attenuated tumor EMT and invasion upon glycolytic inhibition in HNSCC in recent studies implicate glycolysis in facilitating metastasis [67][68].
Given the metabolic rewiring that SCCs undergo to redirect glucose flux into antioxidant generating pathways, lactate from the microenvironment and utilized as a respiratory substrate may serve to maintain energy homeostasis in response to decreased glucose flux into the TCA cycle. Faubert et al. demonstrate that NSCLCs, especially those with highest 18F-FDG uptake and papillary, solid, and squamous histology, preferentially uptake and utilize circulating lactate, not glucose, to fuel the TCA cycle as demonstrated by elevated lactate-to-3PG labeling ratios [69]. Identification of monocarboxylate transporters MCT1 and MCT4 as prognostically significant and generally highly expressed in many SCC subtypes further indicates the essential contribution of lactate transport in SCC oncogenicity [52][70][71][72][73][74]. Rather than primarily a waste product of glycolysis to be eliminated, lactate serves as a critical carbon shuttle and respiratory fuel enabling energetic resilience in metabolically fluctuating conditions and facilitating the diversion of glucose flux into essential anabolic pathways. Thus, the degree to which cancers redirect glucose utilization may determine the propensity by which lactate is catabolized.
Concomitant increases in MCT4, which exports lactate, and glycolysis in cigarette smoke-exposed CAFs and MCT1, which facilitates lactate uptake, and mitochondrial metabolism in HNSCCs demonstrates the essential metabolic coupling driving resistance to cell death and enhanced migration [62]. Furthermore, glucose deprivation in CESCCs, and the associated mitochondrial impairment and increase in ROS, resulted in MCT1 and CD147 upregulation and MCT1-CD147 heterocomplex accumulation, thereby increasing lactate uptake to combat oxidative stress and inducing migration toward more metabolically favorable environments in a lactate-independent manner [75][76]. Thus, MCT1 may not only permit SCCs to tolerate fluctuations in glucose availability by increasing lactate uptake but also dictate tumor invasiveness. However, the non-catabolic respiratory contribution of lactate to macromolecular biosynthetic pathways cannot be discounted, and lactate utilization may be dependent not only on alterations in glucose metabolism but also on oxygen status as lactate oxidation occurs in well-oxygenated regions such as lung tumors, which are well-perfused [72].
Rather than a universally one-dimensional idiosyncrasy of all cancers, the Warburg effect represents a complex, multi-faceted feature that cancers in different contexts utilize uniquely to fulfill various cellular needs, which may help explain the diversity in explanations for this phenomenon. In the SCC context, however, these data taken together reveal the remarkable coordination and adaptation in the uptake and utilization of glucose, glutamine, and lactate to fuel anabolic antioxidant generation while maintaining energy homeostasis, ultimately driving squamous-specific metabolic flexibility.
References
- Ishizumi, T.; McWilliams, A.; MacAulay, C.; Gazdar, A.; Lam, S. Natural history of bronchial preinvasive lesions. Cancer Metastasis Rev. 2010, 29, 5–14.
- Sánchez-Danés, A.; Blanpain, C. Deciphering the cells of origin of squamous cell carcinomas. Nat. Rev. Cancer 2018, 18, 549–561.
- Han, X.; Li, F.; Fang, Z.; Gao, Y.; Li, F.; Fang, R.; Yao, S.; Sun, Y.; Li, L.; Zhang, W.; et al. Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat. Commun. 2014, 5, 3261. Available online: .
- Levin, P.A.; Mayer, M.; Hoskin, S.; Sailors, J.; Oliver, D.H.; Gerber, D.E. Histologic Transformation from Adenocarcinoma to Squamous Cell Carcinoma as a Mechanism of Resistance to EGFR Inhibition. J. Thorac. Oncol. 2015, 10, e86–e88.
- Yan, W.; Wistuba, I.I.; Emmert-Buck, M.R.; Erickson, H.S. Squamous Cell Carcinoma - Similarities and Differences among Anatomical Sites. Am. J. Cancer Res. 2011, 1, 275–300.
- Lapouge, G.; Youssef, K.K.; Vokaer, B.; Achouri, Y.; Michaux, C.; Sotiropoulou, P.A.; Blanpain, C. Identifying the cellular origin of squamous skin tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 7431–7436.
- White, A.C.; Tran, K.; Khuu, J.; Dang, C.; Cui, Y.; Binder, S.W.; Lowry, W.E. Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 7425–7430.
- Ferone, G.; Lee, M.C.; Sage, J.; Berns, A. Cells of origin of lung cancers: Lessons from mouse studies. Genes Dev. 2020, 34, 1017–1032.
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944.
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science 2011, 333, 1157–1160.
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582.
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525.
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322.
- The Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384.
- Cancer Genome Atlas Research Network. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175.
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus Cetuximab for Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2006, 354, 567–578.
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015, 27, 533–546.
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.-R.; Cupissol, D.; et al. Platinum-Based Chemotherapy plus Cetuximab in Head and Neck Cancer. N. Engl. J. Med. 2008, 359, 1116–1127.
- Brown, J.A.; Yonekubo, Y.; Hanson, N.; Sastre-Perona, A.; Basin, A.; Rytlewski, J.A.; Dolgalev, I.; Meehan, S.; Tsirigos, A.; Beronja, S.; et al. TGF-beta-Induced Quiescence Mediates Chemoresistance of Tumor-Propagating Cells in Squamous Cell Carcinoma. Cell Stem Cell 2017, 21, 650–664.e658.
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135.
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496.
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164.
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14.
- Dolado, I.; Swat, A.; Ajenjo, N.; De Vita, G.; Cuadrado, A.; Nebreda, A.R. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell 2007, 11, 191–205.
- Gao, P.; Zhang, H.; Dinavahi, R.; Li, F.; Xiang, Y.; Raman, V.; Bhujwalla, Z.M.; Felsher, D.W.; Cheng, L.; Pevsner, J.; et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 2007, 12, 230–238.
- Kumari, S.; Badana, A.K.; Mohan, G.M.; Naik, G.S.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391.
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203.
- Ju, H.-Q.; Lin, J.-F.; Tian, T.; Xie, D.; Xu, R.-H. NADPH homeostasis in cancer: Functions, mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2020, 5, 231.
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstråhle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514.e422.
- Clifford, G.M.; Smith, J.S.; Plummer, M.; Muñoz, N.; Franceschi, S. Human papillomavirus types in invasive cervical cancer worldwide: A meta-analysis. Br. J. Cancer 2003, 88, 63–73.
- Freedman, N.D.; Abnet, C.C.; Leitzmann, M.F.; Mouw, T.; Subar, A.F.; Hollenbeck, A.R.; Schatzkin, A. A Prospective Study of Tobacco, Alcohol, and the Risk of Esophageal and Gastric Cancer Subtypes. Am. J. Epidemiol. 2007, 165, 1424–1433.
- Madronich, S.; de Gruijl, F.R. Skin cancer and UV radiation. Nature 1993, 366, 23.
- Locasale, J.W.; Cantley, L.C. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011, 14, 443–451.
- Goodwin, J.; Neugent, M.L.; Lee, S.Y.; Choe, J.H.; Choi, H.; Jenkins, D.M.R.; Ruthenborg, R.J.; Robinson, M.W.; Jeong, J.Y.; Wake, M.; et al. The distinct metabolic phenotype of lung squamous cell carcinoma defines selective vulnerability to glycolytic inhibition. Nat. Commun. 2017, 8, 15503.
- Berghmans, T.; Dusart, M.; Paesmans, M.; Hossein-Foucher, C.; Buvat, I.; Castaigne, C.; Scherpereel, A.; Mascaux, C.; Moreau, M.; Roelandts, M.; et al. Primary tumor standardized uptake value (SUVmax) measured on fluorodeoxyglucose positron emission tomography (FDG-PET) is of prognostic value for survival in non-small cell lung cancer (NSCLC): A systematic review and meta-analysis (MA) by the European Lung Cancer Working Party for the IASLC Lung Cancer Staging Project. J. Thorac. Oncol. 2008, 3, 6–12.
- De Geus-Oei, L.F.; van Krieken, J.H.; Aliredjo, R.P.; Krabbe, P.F.; Frielink, C.; Verhagen, A.F.; Boerman, O.C.; Oyen, W.J. Biological correlates of FDG uptake in non-small cell lung cancer. Lung Cancer 2007, 55, 79–87.
- Marom, E.M.; Aloia, T.A.; Moore, M.B.; Hara, M.; Herndon, J.E., 2nd; Harpole, D.H., Jr.; Goodman, P.C.; Patz, E.F., Jr. Correlation of FDG-PET imaging with Glut-1 and Glut-3 expression in early-stage non-small cell lung cancer. Lung Cancer 2001, 33, 99–107.
- Meijer, T.W.; Schuurbiers, O.C.; Kaanders, J.H.; Looijen-Salamon, M.G.; de Geus-Oei, L.F.; Verhagen, A.F.; Lok, J.; van der Heijden, H.F.; Rademakers, S.E.; Span, P.N.; et al. Differences in metabolism between adeno- and squamous cell non-small cell lung carcinomas: Spatial distribution and prognostic value of GLUT1 and MCT4. Lung Cancer 2012, 76, 316–323.
- Hsieh, M.H.; Choe, J.H.; Gadhvi, J.; Kim, Y.J.; Arguez, M.A.; Palmer, M.; Gerold, H.; Nowak, C.; Do, H.; Mazambani, S.; et al. p63 and SOX2 Dictate Glucose Reliance and Metabolic Vulnerabilities in Squamous Cell Carcinomas. Cell Rep. 2019, 28, 1860–1878.e1869.
- De Schutter, H.; Landuyt, W.; Verbeken, E.; Goethals, L.; Hermans, R.; Nuyts, S. The prognostic value of the hypoxia markers CA IX and GLUT 1 and the cytokines VEGF and IL 6 in head and neck squamous cell carcinoma treated by radiotherapy +/- chemotherapy. BMC Cancer 2005, 5, 42.
- Kim, B.H.; Chang, J.H. Differential effect of GLUT1 overexpression on survival and tumor immune microenvironment of human papilloma virus type 16-positive and -negative cervical cancer. Sci. Rep. 2019, 9, 13301.
- Kunkel, M.; Moergel, M.; Stockinger, M.; Jeong, J.H.; Fritz, G.; Lehr, H.A.; Whiteside, T.L. Overexpression of GLUT-1 is associated with resistance to radiotherapy and adverse prognosis in squamous cell carcinoma of the oral cavity. Oral Oncol. 2007, 43, 796–803.
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302.
- Peng, L.; Linghu, R.; Chen, D.; Yang, J.; Kou, X.; Wang, X.-Z.; Hu, Y.; Jiang, Y.-Z.; Yang, J. Inhibition of glutathione metabolism attenuates esophageal cancer progression. Exp. Mol. Med. 2017, 49, e318.
- Sobhakumari, A.; Love-Homan, L.; Fletcher, E.V.; Martin, S.M.; Parsons, A.D.; Spitz, D.R.; Knudson, C.M.; Simons, A.L. Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS ONE 2012, 7, e48175.
- Mele, L.; Paino, F.; Papaccio, F.; Regad, T.; Boocock, D.; Stiuso, P.; Lombardi, A.; Liccardo, D.; Aquino, G.; Barbieri, A.; et al. A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo. Cell Death Dis. 2018, 9, 572.
- Sellers, K.; Allen, T.D.; Bousamra, M.; Tan, J.; Méndez-Lucas, A.; Lin, W.; Bah, N.; Chernyavskaya, Y.; MacRae, J.I.; Higashi, R.M.; et al. Metabolic reprogramming and Notch activity distinguish between non-small cell lung cancer subtypes. Br. J. Cancer 2019, 121, 51–64.
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000, 14, 34–44.
- Veldman, T.; Liu, X.; Yuan, H.; Schlegel, R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc. Natl. Acad. Sci. USA 2003, 100, 8211–8216.
- Zwerschke, W.; Mazurek, S.; Massimi, P.; Banks, L.; Eigenbrodt, E.; Jansen-Dürr, P. Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc. Natl. Acad. Sci. USA 1999, 96, 1291–1296.
- Zeng, Q.; Chen, J.; Li, Y.; Werle, K.D.; Zhao, R.X.; Quan, C.S.; Wang, Y.S.; Zhai, Y.X.; Wang, J.W.; Youssef, M.; et al. LKB1 inhibits HPV-associated cancer progression by targeting cellular metabolism. Oncogene 2017, 36, 1245–1255.
- Fleming, J.C.; Woo, J.; Moutasim, K.; Mellone, M.; Frampton, S.J.; Mead, A.; Ahmed, W.; Wood, O.; Robinson, H.; Ward, M.; et al. HPV, tumour metabolism and novel target identification in head and neck squamous cell carcinoma. Br. J. Cancer 2019, 120, 356–367.
- Jung, Y.S.; Najy, A.J.; Huang, W.; Sethi, S.; Snyder, M.; Sakr, W.; Dyson, G.; Hüttemann, M.; Lee, I.; Ali-Fehmi, R.; et al. HPV-associated differential regulation of tumor metabolism in oropharyngeal head and neck cancer. Oncotarget 2017, 8, 51530–51541.
- Prusinkiewicz, M.A.; Gameiro, S.F.; Ghasemi, F.; Dodge, M.J.; Zeng, P.Y.F.; Maekebay, H.; Barrett, J.W.; Nichols, A.C.; Mymryk, J.S. Survival-Associated Metabolic Genes in Human Papillomavirus-Positive Head and Neck Cancers. Cancers 2020, 12, 253.
- Flores, A.; Sandoval-Gonzalez, S.; Takahashi, R.; Krall, A.; Sathe, L.; Wei, L.; Radu, C.; Joly, J.H.; Graham, N.A.; Christofk, H.R.; et al. Increased lactate dehydrogenase activity is dispensable in squamous carcinoma cells of origin. Nat. Commun. 2019, 10, 91.
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid transporters in cancer and their relevance to "glutamine addiction": Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788.
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padró, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; van’t Veer, L.J.; et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 2013, 24, 450–465.
- Qie, S.; Yoshida, A.; Parnham, S.; Oleinik, N.; Beeson, G.C.; Beeson, C.C.; Ogretmen, B.; Bass, A.J.; Wong, K.-K.; Rustgi, A.K.; et al. Targeting glutamine-addiction and overcoming CDK4/6 inhibitor resistance in human esophageal squamous cell carcinoma. Nat. Commun. 2019, 10, 1296.
- Zhang, Z.; Liu, R.; Shuai, Y.; Huang, Y.; Jin, R.; Wang, X.; Luo, J. ASCT2 (SLC1A5)-dependent glutamine uptake is involved in the progression of head and neck squamous cell carcinoma. Br. J. Cancer 2020, 122, 82–93.
- Luo, Y.; Li, W.; Ling, Z.; Hu, Q.; Fan, Z.; Cheng, B.; Tao, X. ASCT2 overexpression is associated with poor survival of OSCC patients and ASCT2 knockdown inhibited growth of glutamine-addicted OSCC cells. Cancer Med. 2020, 9, 3489–3499.
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Fishbein, M.C.; Braas, D.; Go, J.; Graeber, T.G.; Parlati, F.; Demo, S.; Li, R.; et al. The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma. Cancer Cell 2018, 33, 905–921.e905.
- Domingo-Vidal, M.; Whitaker-Menezes, D.; Martos-Rus, C.; Tassone, P.; Snyder, C.M.; Tuluc, M.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Cigarette Smoke Induces Metabolic Reprogramming of the Tumor Stroma in Head and Neck Squamous Cell Carcinoma. Mol. Cancer Res. 2019.
- Kumar, D.; New, J.; Vishwakarma, V.; Joshi, R.; Enders, J.; Lin, F.; Dasari, S.; Gutierrez, W.R.; Leef, G.; Ponnurangam, S.; et al. Cancer-Associated Fibroblasts Drive Glycolysis in a Targetable Signaling Loop Implicated in Head and Neck Squamous Cell Carcinoma Progression. Cancer Res. 2018, 78, 3769–3782.
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140.
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535.
- Rohani, N.; Hao, L.; Alexis, M.S.; Joughin, B.A.; Krismer, K.; Moufarrej, M.N.; Soltis, A.R.; Lauffenburger, D.A.; Yaffe, M.B.; Burge, C.B.; et al. Acidification of Tumor at Stromal Boundaries Drives Transcriptome Alterations Associated with Aggressive Phenotypes. Cancer Res. 2019, 79, 1952.
- Li, H.M.; Yang, J.G.; Liu, Z.J.; Wang, W.M.; Yu, Z.L.; Ren, J.G.; Chen, G.; Zhang, W.; Jia, J. Blockage of glycolysis by targeting PFKFB3 suppresses tumor growth and metastasis in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 7.
- Xu, Q.; Zhang, Q.; Ishida, Y.; Hajjar, S.; Tang, X.; Shi, H.; Dang, C.V.; Le, A.D. EGF induces epithelial-mesenchymal transition and cancer stem-like cell properties in human oral cancer cells via promoting Warburg effect. Oncotarget 2017, 8, 9557–9571.
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e359.
- Zhu, J.; Wu, Y.N.; Zhang, W.; Zhang, X.M.; Ding, X.; Li, H.Q.; Geng, M.; Xie, Z.Q.; Wu, H.M. Monocarboxylate transporter 4 facilitates cell proliferation and migration and is associated with poor prognosis in oral squamous cell carcinoma patients. PLoS ONE 2014, 9, e87904.
- Sweeny, L.; Dean, N.R.; Frederick, J.W.; Magnuson, J.S.; Carroll, W.R.; Desmond, R.A.; Rosenthal, E.L. CD147 expression in advanced cutaneous squamous cell carcinoma. J. Cutan. Pathol. 2012, 39, 603–609.
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942.
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66.
- Izumi, H.; Takahashi, M.; Uramoto, H.; Nakayama, Y.; Oyama, T.; Wang, K.-Y.; Sasaguri, Y.; Nishizawa, S.; Kohno, K. Monocarboxylate transporters 1 and 4 are involved in the invasion activity of human lung cancer cells. Cancer Sci. 2011, 102, 1007–1013.
- De Saedeleer, C.J.; Porporato, P.E.; Copetti, T.; Pérez-Escuredo, J.; Payen, V.L.; Brisson, L.; Feron, O.; Sonveaux, P. Glucose deprivation increases monocarboxylate transporter 1 (MCT1) expression and MCT1-dependent tumor cell migration. Oncogene 2014, 33, 4060–4068.
- Payen, V.L.; Hsu, M.Y.; Rädecke, K.S.; Wyart, E.; Vazeille, T.; Bouzin, C.; Porporato, P.E.; Sonveaux, P. Monocarboxylate Transporter MCT1 Promotes Tumor Metastasis Independently of Its Activity as a Lactate Transporter. Cancer Res. 2017, 77, 5591–5601.

