 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Huey-Kang Sytwu | + 1521 word(s) | 1521 | 2020-05-13 09:02:12 | | | |
| 2 | Catherine Yang | -19 word(s) | 1502 | 2020-10-28 07:37:31 | | |
Video Upload Options
Inflammatory bowel disease (IBD) is a chronic disorder manifested as Crohn’s disease (CD) and ulcerative colitis (UC) characterized by intestinal inflammation and involves a dysregulated immune response against commensal microbiota through the activation of CD4 T helper cells. T helper cell differentiation to effector or regulatory phenotypes is controlled by cytokine networks and transcriptional regulators. Distinct polarized T helper cells are able to alter their phenotypes to adapt to diverse and fluctuating physiological environments. T helper cells exhibit intrinsic instability and flexibility to express cytokines of other lineages or transdifferentiate from one T helper cell type to another in response to various perturbations from physiological cytokine milieu as a means of promoting local immunity in response to injury or ensure tissue homeostasis. Furthermore, functional plasticity and diversity of T helper cells are associated with pathogenicity and are critical for immune homeostasis and prevention of autoimmunity. In this review, we provide deeper insights into the combinatorial extrinsic and intrinsic signals that control plasticity and transdifferentiation of T helper cells and also highlight the potential of exploiting the genetic reprogramming plasticity of T helper cells in the treatment of IBD.
1. Introduction
T cell lineage commitment to a single fixed Th phenotype has been challenged by increasing evidence of functional plasticity within CD4+ T cell subsets with the ability to change between helper phenotypes, or helper and regulatory functions. The capacity for T cell lineages to switch between patterns of cytokine secretion and effector functions from one lineage to another under tissue environmental cues is thought to be reprogrammed by the expression of transcription factors characteristic of opposing lineages. Notably, recent evidence on conversion among polarized T helper cells highlighted the significance of Th cell plasticity and transdifferentiation in the pathogenesis of IBD [1][2][3][4][5].
2. Th1 Plasticity
Although the well-defined Th1 lineages are thought of as terminally committed effector cells which exhibit low levels of plasticity in changing their phenotype, fully differentiated myelin oligodendrocyte glycoprotein (MOG)-specific Th1 clones are still capable of producing IL-17 upon superantigen stimulation [6]. In addition, human Th1 cells exposed to Th17-polarizing conditions undergo lineage conversion and exhibit a Th1/17 phenotype by upregulating IL-17 upon enforced expression of RORC2 but without losing expression of IFN-γ or TBX21 [7]. Retinoic acid (RA) signaling was reported to be important for limiting Th1 cell conversion into Th17 effector cells and for preventing pathogenic Th17 responses in vivo through maintaining stable expression of Th1 lineage-specific genes, as well as repressing genes that instruct Th17-cell fate [8]. IFN-γ-producing Th1 effector cells specific for an intestinal microbial antigen, CBir1 flagellin, induced colitis in Rag-/- mice after adoptive transfer and acquired IL-17-producing capacities in the gut. Th1 conversion into Th17 cells is regulated by TGF-β and IL-6, and mediated by TGF-β induction of Runx1. TGF-β enhances accessibility of Runx1 binding to RORγt and IL-17 promoters in Th1 cells, supporting a possible mechanism for Th1 to Th17 cell transdifferentiation under inflammatory conditions in the intestine [9]. Another report uncovered a small ubiquitin-related modifier (SUMO)ylation-based mechanism that controls Th1/Th17 plasticity, SUMOylation-deficient Wiskott-Aldrich syndrome protein (WASp), which favors ectopic development of the Th17-like phenotype under Th1-skewing conditions, while SUMOylated nuclear-WASp serves as a transcriptional promoter-specific coactivator, which is essential in Th1 gene transcription [10]. However, IFN-γ-producing cells tend not to develop into Tregs and inhibit the generation of Tregs in vivo, suggesting limited plasticity of Th1 populations to Tregs [11]. Nevertheless, upon short-term culture in vitro, effector CD4+CD25-CD127+ T cells from human peripheral blood could convert into T cells with regulatory activity while expressing a panel of common Treg markers, including FOXP3, CD25, GITR, HLA-DR, and CTLA-4, in parallel with the Th1-specific transcription factor T-bet and concomitantly secreting IFN-γ [12]. These data reveal a unique regulatory network governing maintenance and plasticity of Th1-cell fate and define an additional pathway for the development of Th17 cells.
3. Th17 and Treg Imbalance Paradigms
A loss of homeostasis between regulatory and inflammatory CD4+ T cell populations results in subsequent intestinal tissue damage, while increased Th1, Th2, Th9, and Th17 responses and reduced Treg and Tr1 responses were suggested to play a role in IBD pathophysiology [4]. Although Th17 and Treg cells are two distinct T cell subsets with opposite effects on immune functions, the reciprocal differentiation of Th17 cells and Treg cells is closely related and plays a critical role in both the pathogenesis and resolution of IBD. Tregs were capable of suppressing Th17-mediated colitis in an adoptive transfer model of colitis [13]. Decreased Tregs, increased Th17 cells, and a significant decrease in the Treg/Th17 ratio were observed in the peripheral blood of IBD and pediatric IBD patients; this reduced ratio is associated with serum IL-6 and IL-23 levels, suggesting that the Th17/Treg cell balance plays an important role in the development and maintenance of inflammation [14][15][16]. In particular, disordered regulation of Th17 and Treg cells in IBD is caused by a disruption in the chemokines and, consequently, their cognate receptors, thereby disturbing mucosal homeostasis [17][18]. IL-6 is a proinflammatory cytokine favoring Th17 differentiation, whereas SMAD4 is only involved in Foxp3 upregulation. TGF-β signaling is required for both Foxp3 and IL-17 induction. However, IL-6 is crucial for the balance between pathogenic Th17 and protective Treg by overcoming the suppressive effect of Foxp3 (Figure 1). Moreover, IL-6 and IL-1 induce genetic reprogramming in Treg cells. Foxp3 antagonizes RORγt to inhibit Th17 differentiation while STAT3 downregulates Foxp3 expression and is critical for IL-17 expression in Treg cells [19]. Further, environmental cues including the gut microbita and dietary components are involved in regulating Th17/Treg cell imbalance paradigms. All-trans retinoic acid (ATRA), the active derivative of vitamin A, inhibits differentiation of Th17 cells and also induces Treg differentiation in colon biopsies of patients with UC in vitro and in TNBS-induced murine colitis, restoring the Th17/Treg balance [20]. A high salt diet stimulated the intestinal Th17 response but inhibited the function of Treg cells in TNBS-induced mice colitis [21]. Segmented filamentous bacteria modulate the homeostatic plasticity of T helper and T regulatory cells and coordinate the intestinal T cell profile [22]. A plethora of therapeutic strategies for IBD regulation demonstrated that DSS-induced mouse colitis was ameliorated via regulating the balance between Th17 and Treg cell to control intestinal inflammation [23][24][25][26] Moreover, impaired delivery of IL-15 to CD4+ T cells and CD4+ T cells deprived of IL-15 can trigger intestinal inflammation by accumulation of Th1/Th17 cells and fine-tune the balance between Treg and Th17 cells by downmodulating Foxp3 expression and enhancing RORγt expression [27]. Furthermore, growing evidence reports remarkable plasticity between the Th17 and Treg cells reflected in the capacity of differentiated effectors cells to be reprogrammed among Th17 and Treg lineages [28]. A crucial need is emerging to understand the complexity of the specificity and plasticity of effector Th17 and suppressive Treg cells, which are central to the pathogenesis of IBD.
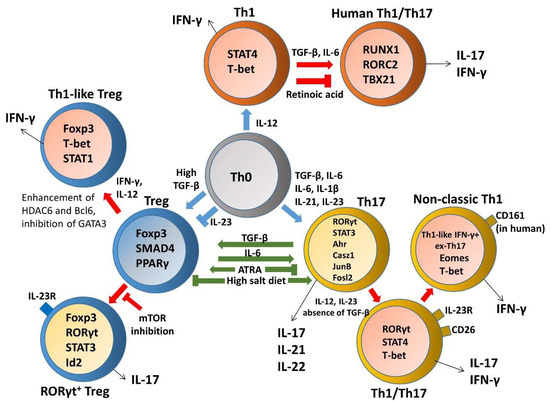
Figure 1. The plasticity and transdifferentiation of Th1, Th17, and Treg cells. Cytokine networks influencing T cell development are indicated in blue arrows and extrinsic signals that control plasticity are shown in red arrows. Cytokine production by distinct T helper cells is indicated in black arrows and components involved in regulating Th17/Treg cell imbalance paradigms are indicated in green arrows.
4. Th17 Plasticity
In contrast to the classic Th1 and Th2 cells, studies of Th17 differentiation elucidated that developmental flexibility and heterogeneity allows them to acquire cytotoxic activity or regulatory phenotype in the periphery in response to additional environmental challenges within the tissue [29]. Since the initial finding that TGF-β is required for both Th17 and Treg cell differentiation, plasticity between Treg and Th17 cell programs was shown to be reciprocally regulated. Subsequent findings revealed that differentiated Th17 subsets displayed a high grade of plasticity to produce predominantly IFN-γ in terminal differentiation, which is not an endpoint of T cell development, implying substantial overlap with conventional Th1 cells [30][31]. It became apparent that cytokine signals and transcriptional factors determine the fate of Th17 cell plasticity, leading them to either fully acquire their pathogenicity or divert toward a regulatory fate. Evidence from animal and human IBD studies highlighted an important role of T cell plasticity in the regulation of mucosal homeostasis and inflammation, such as Th1–Th17 and Th17–Treg axes. Moreover, increased numbers of transdifferentiated T cell populations developed in IBD patients displayed a high grade of plasticity [32][33][34]. Taken together, deeper insights into plasticity in inflammatory conditions could contribute to the design of therapeutic strategies for IBD by reprogramming the fate of Th17 in the gut.
5. Treg Plasticity
Recent studies suggested FoxP3+ Treg cells are a heterogeneous population consisting of a committed Treg lineage and an uncommitted subpopulation with a high grade of plasticity, allowing them to their cell fate to various effector T cell phenotypes as a functional adaptation to inflammation and as a critical factor for autoimmune disease. However, whether post-conversion Tregs maintain stable Foxp3 expression as well as suppressive function or lose Foxp3 expression remains under debate [35][36][37].
References
- Loo, T.T.; Gao, Y.; Lazarevic, V. Transcriptional regulation of CD4+ TH cells that mediate tissue inflammation. J. Leukoc. Biol. 2018, 104, 1069–1085.
- Rowell, E.; Wilson, C.B. Programming perpetual T helper cell plasticity. Immunity 2009, 30, 7–9.
- Wei, G.; Wei, L.; Zhu, J.; Zang, C.; Hu-Li, J.; Yao, Z.; Cui, K.; Kanno, Y.; Roh, T.Y.; Watford, W.T.; et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 2009, 30, 155–167.
- Lee, Y.K.; Turner, H.; Maynard, C.L.; Oliver, J.R.; Chen, D.; Elson, C.O.; Weaver, C.T. Late developmental plasticity in the T helper 17 lineage. Immunity 2009, 30, 92–107.
- Tindemans, I.; Joosse, M.E.; Samsom, J.N. Dissecting the Heterogeneity in T-Cell Mediated Inflammation in IBD. Cells 2020, 9, 110.
- Yomogida, K.; Chou, Y.K.; Chu, C.Q. Superantigens induce IL-17 production from polarized Th1 clones. Cytokine 2013, 63, 6–9.
- Cohen, C.J.; Crome, S.Q.; MacDonald, K.G.; Dai, E.L.; Mager, D.L.; Levings, M.K. Human Th1 and Th17 cells exhibit epigenetic stability at signature cytokine and transcription factor loci. J. Immunol. 2011, 187, 5615–5626.
- Brown, C.C.; Esterhazy, D.; Sarde, A.; London, M.; Pullabhatla, V.; Osma-Garcia, I.; Al-Bader, R.; Ortiz, C.; Elgueta, R.; Arno, M.; et al. Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 2015, 42, 499–511.
- Liu, H.P.; Cao, A.T.; Feng, T.; Li, Q.; Zhang, W.; Yao, S.; Dann, S.M.; Elson, C.O.; Cong, Y. TGF-beta converts Th1 cells into Th17 cells through stimulation of Runx1 expression. Eur. J. Immunol. 2015, 45, 1010–1018.
- Sarkar, K.; Sadhukhan, S.; Han, S.-S.; Vyas, Y.M. SUMOylation-disrupting WAS mutation converts WASp from a transcriptional activator to a repressor of NF-κB response genes in T cells. Blood 2015, 126, 1670–1682.
- Caretto, D.; Katzman, S.D.; Villarino, A.V.; Gallo, E.; Abbas, A.K. Cutting edge: The Th1 response inhibits the generation of peripheral regulatory T cells. J. Immunol. 2010, 184, 30–34.
- Venigalla, R.K.; Guttikonda, P.J.; Eckstein, V.; Ho, A.D.; Sertel, S.; Lorenz, H.M.; Tretter, T. Identification of a human Th1-like IFNgamma-secreting Treg subtype deriving from effector T cells. J. Autoimmun. 2012, 39, 377–387.
- Ogino, H.; Nakamura, K.; Ihara, E.; Akiho, H.; Takayanagi, R. CD4+CD25+ regulatory T cells suppress Th17-responses in an experimental colitis model. Dig. Dis. Sci. 2011, 56, 376–386.
- Eastaff-Leung, N.; Mabarrack, N.; Barbour, A.; Cummins, A.; Barry, S. Foxp3+ regulatory T cells, Th17 effector cells, and cytokine environment in inflammatory bowel disease. J. Clin. Immunol. 2010, 30, 80–89.
- Geng, X.; Xue, J. Expression of Treg/Th17 cells as well as related cytokines in patients with inflammatory bowel disease. Pak. J. Med. Sci. 2016, 32, 1164–1168.
- Zhu, X.M.; Shi, Y.Z.; Cheng, M.; Wang, D.F.; Fan, J.F. Serum IL-6, IL-23 profile and Treg/Th17 peripheral cell populations in pediatric patients with inflammatory bowel disease. Pharmazie 2017, 72, 4.
- Lee, A.Y.; Eri, R.; Lyons, A.B.; Grimm, M.C.; Korner, H. CC Chemokine Ligand 20 and Its Cognate Receptor CCR6 in Mucosal T Cell Immunology and Inflammatory Bowel Disease: Odd Couple or Axis of Evil? Front. Immunol. 2013, 4, 194.
- McNamee, E.N.; Masterson, J.C.; Veny, M.; Collins, C.B.; Jedlicka, P.; Byrne, F.R.; Ng, G.Y.; Rivera-Nieves, J. Chemokine receptor CCR7 regulates the intestinal TH1/TH17/Treg balance during Crohn’s-like murine ileitis. J. Leukoc. Biol. 2015, 97, 1011–1022.
- Yang, X.O.; Nurieva, R.; Martinez, G.J.; Kang, H.S.; Chung, Y.; Pappu, B.P.; Shah, B.; Chang, S.H.; Schluns, K.S.; Watowich, S.S.; et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 2008, 29, 44–56.
- Bai, A.; Lu, N.; Guo, Y.; Liu, Z.; Chen, J.; Peng, Z. All-trans retinoic acid down-regulates inflammatory responses by shifting the Treg/Th17 profile in human ulcerative and murine colitis. J. Leukoc. Biol. 2009, 86, 959–969.
- Wei, Y.; Lu, C.; Chen, J.; Cui, G.; Wang, L.; Yu, T.; Yang, Y.; Wu, W.; Ding, Y.; Li, L.; et al. High salt diet stimulates gut Th17 response and exacerbates TNBS-induced colitis in mice. Oncotarget 2017, 8, 70–82.
- Gaboriau-Routhiau, V.; Rakotobe, S.; Lecuyer, E.; Mulder, I.; Lan, A.; Bridonneau, C.; Rochet, V.; Pisi, A.; De Paepe, M.; Brandi, G.; et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 2009, 31, 677–689.
- Wang, J.; Tian, M.; Li, W.; Hao, F. Preventative delivery of IL-35 by Lactococcus lactis ameliorates DSS-induced colitis in mice. Appl. Microbiol. Biotechnol. 2019, 103, 7931–7941.
- Higashiyama, M.; Hokari, R.; Hozumi, H.; Kurihara, C.; Ueda, T.; Watanabe, C.; Tomita, K.; Nakamura, M.; Komoto, S.; Okada, Y.; et al. HIF-1 in T cells ameliorated dextran sodium sulfate-induced murine colitis. J. Leukoc. Biol. 2012, 91, 901–909.
- Zhang, L.; Zhang, Y.; Zhong, W.; Di, C.; Lin, X.; Xia, Z. Heme oxygenase-1 ameliorates dextran sulfate sodium-induced acute murine colitis by regulating Th17/Treg cell balance. J. Biol. Chem. 2014, 289, 26847–26858.
- Hu, S.; Chen, M.; Wang, Y.; Wang, Z.; Pei, Y.; Fan, R.; Liu, X.; Wang, L.; Zhou, J.; Zheng, S.; et al. mTOR Inhibition Attenuates Dextran Sulfate Sodium-Induced Colitis by Suppressing T Cell Proliferation and Balancing TH1/TH17/Treg Profile. PLoS ONE 2016, 11, e0154564.
- Tosiek, M.J.; Fiette, L.; El Daker, S.; Eberl, G.; Freitas, A.A. IL-15-dependent balance between Foxp3 and RORgammat expression impacts inflammatory bowel disease. Nat. Commun. 2016, 7, 10888.
- Diller, M.L.; Kudchadkar, R.R.; Delman, K.A.; Lawson, D.H.; Ford, M.L. Balancing Inflammation: The Link between Th17 and Regulatory T Cells. Mediat. Inflamm. 2016, 2016, 6309219.
- Muranski, P.; Restifo, N.P. Essentials of Th17 cell commitment and plasticity. Blood 2013, 121, 2402–2414.
- Basu, R.; Hatton, R.D.; Weaver, C.T. The Th17 family: Flexibility follows function. Immunol. Rev. 2013, 252, 89–103.
- Lee, Y.K.; Mukasa, R.; Hatton, R.D.; Weaver, C.T. Developmental plasticity of Th17 and Treg cells. Curr. Opin. Immunol. 2009, 21, 274–280.
- Ueno, A.; Jeffery, L.; Kobayashi, T.; Hibi, T.; Ghosh, S.; Jijon, H. Th17 plasticity and its relevance to inflammatory bowel disease. J. Autoimmun. 2018, 87, 38–49.
- Ueno, A.; Ghosh, A.; Hung, D.; Li, J.; Jijon, H. Th17 plasticity and its changes associated with inflammatory bowel disease. World J. Gastroenterol. 2015, 21, 12283–12295.
- Rossi, M.; Bot, A. The Th17 cell population and the immune homeostasis of the gastrointestinal tract. Int. Rev. Immunol. 2013, 32, 471–474.
- Hori, S. Lineage Stability and Phenotypic Plasticity of Foxp3⁺ Regulatory T Cells. Immunol. Rev. 2014, 259, 13.
- Komatsu, N.; Mariotti-Ferrandiz, M.E.; Wang, Y.; Malissen, B.; Waldmann, H.; Hori, S. Heterogeneity of natural Foxp3+ T cells: A committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc. Natl. Acad. Sci. USA 2009, 106, 1903–1908.
- Esposito, M.; Ruffini, F.; Bergami, A.; Garzetti, L.; Borsellino, G.; Battistini, L.; Martino, G.; Furlan, R. IL-17- and IFN-gamma-secreting Foxp3+ T cells infiltrate the target tissue in experimental autoimmunity. J. Immunol. 2010, 185, 7467–7473.

