 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Diego Angosto Bazarra | + 2858 word(s) | 2858 | 2021-03-24 03:49:46 | | | |
| 2 | Bruce Ren | -21 word(s) | 2837 | 2021-03-25 02:42:01 | | | | |
| 3 | Bruce Ren | -20 word(s) | 2838 | 2021-03-25 02:50:27 | | |
Video Upload Options
Inflammasomes are immune cytosolic oligomers involved in the initiation and progression of multiple pathologies and diseases. The tight regulation of these immune sensors is necessary to control an optimal inflammatory response and recover organism homeostasis. Prolonged activation of inflammasomes result in the development of chronic inflammatory diseases, and the use of small drug-like inhibitory molecules are emerging as promising anti-inflammatory therapies.
1. Introduction
Nearly two decades ago, an intracellular caspase-activating multiprotein complex was named as the “inflammasome” [1]. Inflammasomes can be defined as cytosolic-multimeric-high molecular weight protein oligomers that are formed in response to different intrinsic and/or external cellular stimuli including pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs) or homeostasis-altering molecular processes (HAMPs) (Table 1) [2][3][4]. The inflammasomes are classified and named according to the sensor protein that triggers its activation, the different inflammasome sensors share structural motifs and could belong to the nucleotide-binding domain and leucine-rich-containing family of receptors (NLRs), the absent in melanoma 2-like receptors (ALRs) and pyrin (Figure 1) [5][6].
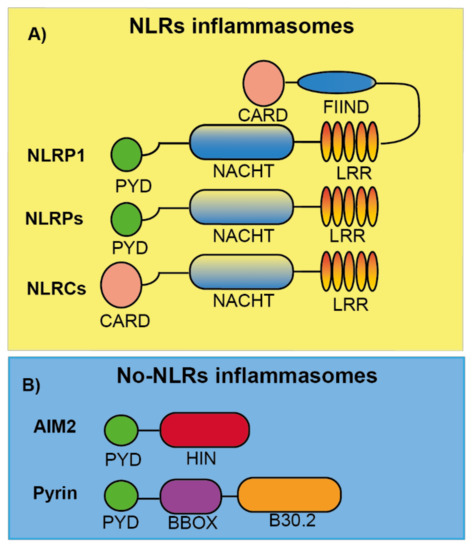
Figure 1. Schematic representation of the different inflammasome sensor proteins. (A) Nod like receptor (NLR) proteins are formed by either a PYD (NLRP) or CARD (NLRC) domain at the N-terminal position follow by a central domain (NACHT), responsible for NLR oligomerization, and a C-terminal leucine-rich repeat domain (LRR). For NLRP1, a FIIND and CARD domain are in the C-terminal position after the LRR domain. (B) AIM2 and Pyrin are no-NLR inflammasome sensors with a PYD domain at the N-terminal. AIM2 also presents a HIN domain responsible of DNA binding. Pyrin is also formed by a central coiled BBOX domain and a C-terminal B30.2/SPRY domain.
Table 1. PAMPs (pathogen-associated molecular patterns) and DAMPs (damage-associated molecular patterns) responsible for inflammasome activation.
| PAMPs | Lipopolysaccharide (LPS), Bacterial lipoproteins and lipopeptides, Porins, Peptidoglycan, Lipoteichoic acids, Mannose-rich glycans, Flagellin, Bacterial and viral nucleic acid, Single and double-stranded viral RNA, Glycolipids, Zymosan, lipids from microbial membranes. |
| DAMPs | Adenosin-5′-phosphate (ATP), Adenosine monophosphate (AMP), Adenosine, High mobility group box 1 (HMGB1), Double-stranded DNA, Chromatin, RNA, Monosodium urate, oxidation products, Heat shock proteins, Defensins, β-amyloid, Calcium binding proteins, Mitochondrial DNA, Matrix proteins, Hyaluronic acid, Collagen peptides, Integrins. |
To engage the inflammasomes, these sensors homo-oligomerize and directly recruit the zymogen of caspase-1 through homotypic interactions or through the adaptor molecule apoptosis-associated speck-like protein containing a caspase recruitment and activation domain (ASC) as occurs in most of the inflammasomes. ASC oligomerization to inflammasome sensors result in one of the hallmarks of inflammasome activation, the formation of a large ASC oligomer named “ASC speck” [7][8]. The zymogen of caspase-1 is activated within the inflammasome due to proximity induced autocatalytic cleavage of two precursors subunits [9]. Once fully activated, caspase-1 cleaves different protein substrates in the cell that include the consensus sequence YVHD/FESD [10]. Among the different caspase-1 substrates, we found the pro-inflammatory cytokines interleukin (IL)-1β and IL-18, and the protein gasdermin D (GSDMD) [11]. After caspase-1 processing, the generated N-terminal fragment of GSDMD binds to negatively charge lipids of the plasma membrane inner bilayer and oligomerizes producing a pore of about 10-12 nm, from where the mature IL-1β and IL-18, as well as other intracellular content, is released from the cell [11]. If GSDMD pores are not efficiently removed from the plasma membrane, a specific type of cell death, called pyroptosis, is produced (Figure 2) [11][12].
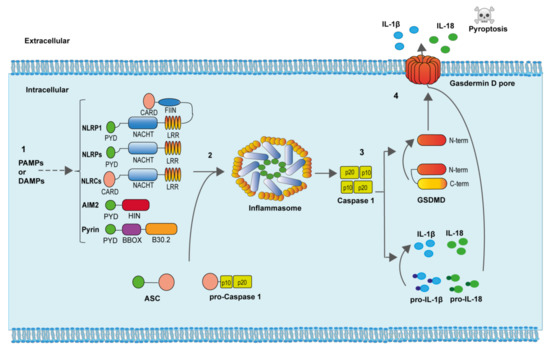
Figure 2. Inflammasome activation. (1) Specific PAMPs and DAMPs are able to induce both the expression and oligomerization of the different inflammasome sensor proteins. (2) The adaptor ASC protein oligomerizes within the inflammasome and recruits pro-caspase-1, favoring caspase-1 autoactivation. (3) Active caspase-1 cleaves the precursors of the pro-inflammatory cytokines IL-1β and IL-18, as well as the protein GSDMD. The N-terminal portion of GSDMD binds and produce pores in the plasma membrane. (4) The active pro-inflammatory cytokines IL-1β and IL-18, as well as diverse intracellular content, is released through GSDMD pores. Increasing formation of GSDMD pores produces cell swelling and pyroptotic cell death.
Inflammasomes are important components of host defense and are able to induce an effective immune response. A controlled activation of inflammasomes is critical to obtain an optimal response against different insults, including infections. A dysregulation of inflammasome activation is associated with different pathologies, such as neurodegenerative diseases (like Alzheimer or multiple sclerosis), metabolic diseases (like type 2 diabetes or non-alcoholic steatohepatitis), autoimmune diseases (like rheumatoid arthritis or gout), the rejection of allogeneic transplants, and autoinflammatory syndromes where specific mutations in different inflammasome sensors encoding genes are responsible of an exacerbated inflammasome activation [13][14][15][16][17][18][19][20][21]. Due to the important implication of the inflammasomes in different pathologies, the identification of specific inflammasome inhibitors is a promising growing field to manage different diseases that currently lack effective therapies [22]. Different techniques are needed to reveal the mechanism of action of inflammasome inhibitors, and this review aims to summarize them.
2. NLR Inflammasomes
NLR inflammasomes can be grouped into two major families, the NLRPs and the NLRCs, depending on whether the N-terminal presents a pyrin (PYD) or caspase recruitment and activation (CARD) domain, respectively (Figure 1A) [6]. In humans, there are 14 NLRP and 5 NLRC identified [23]. Both share some features as a C-terminal leucine-rich repeat domain (LRR) and a central domain responsible for NLR oligomerization found in different plant and animal proteins named NACHT (neuronal apoptosis inhibitory protein NAIP, major histocompatibility class II transcription activator CIITA, incompatibility locus protein from Podospora anserina HET-E, and telomerase-associated protein TP1) [23]. Several proteins of this family have been well characterized to form inflammasomes, among them we found the NLRP1, NLRP3, NLRP6, and NLRC4 inflammasomes, while other NLRs members remain to be studied if they are able to form inflammasomes in response to specific stimuli.
2.1. NLRP1 Inflammasome
NLRP1 was the first inflammasome to be described [1]. The NLRP1 has different domains: PYD, NACHT, LRR, a function to find (FIIND) domain, and a CARD domain (Figure 1A). In mice, NLRP1 presents three different isoforms: NLRP1a, NLRP1b, and NLRP1c, and differentially from the single human NLRP1, all of them lack the PYD domain. NLRP1b has been widely studied and is activated by anthrax lethal toxin that is produced by Bacillus anthracis. Anthrax lethal toxin is formed by a protective antigen and a lethal factor with metalloprotease activity responsible for the specific cleavage inside the FIIND domain of NLRP1b. This cleavage is necessary to activate the NLRP1 inflammasome, since the NLRP1b N-terminal fragment is degraded by the proteasome, liberating the C-terminal CARD domain responsible for caspase-1 activation [24][25][26][27] (Table 2). Therefore, the NLRP1b needs the processing of its FIIND domain to produce an active inflammasome, either by external stimulations or autoprocessing [28]. NLRP1b activation is important for parasite clearance and acquiring specific protection against Toxoplasma gondii [29]. This role has also been found for the human NLRP1, where a loss of NLRP1 in monocytic cells fosters Toxoplasma infectivity [30]. However, the specific ligand or activation for the human NLRP1 activation has not been elucidated yet [31]. In humans, specific mutations of NLRP1 gene are related with vitiligo, Addison’s disease, skin inflammation, and cancer susceptibility [32][33][34], but more studies are necessary to shed light into the molecular mechanism of NLRP1 induced pathology. So far, there are not any reported NLRP1 specific inhibitors that could be important to treat the impact of the diseases where NLRP1 is involved.
Table 2. Activators of Different Inflammasomes.
| Inflammasome | Activators |
|---|---|
| NLRP1b | B. anthracis lethal toxin, T. gondii, Muramyl dipeptide |
| NLRP3 | C. albicans, S. cerevisiae, S. aureus, L. monocytogenes, Influenza virus, Sendai virus, Adenovirus, Bacterial pore-forming toxins, Hemozoin, Silica, Asbestos, Ultra violet (U.V.), ATP, Glucose, Monosodium urate (MSU) crystals, Calcium pyrophosphate dehydrate, β-amyloid, Aluminum particles (Alum), Imiquimod, Hyaluronan, ROS, Cholesterol crystals, Cell swelling |
| NLRC4 | Cytosolic Flagellin, Type III secretion system (T3SS) rod protein, T3SS needle complex protein PrgI |
| NLRP6 | Lipoteichoic acid, Gut metabolites, Microbial RNA, LPS |
| AIM2 | dsDNA from DNA viruses or cytosolic bacteria |
| Pyrin | Bacterial toxins-inducing Rho guanosine triphosphatase (Rho GTPase) inhibition, such as the ones from C. difficile, H. somni, V. parahaemolyticus, or Y. pestis |
2.2. NLRP3 Inflammasome
This is the most studied inflammasome due to the wide number of pathologies where it is implicated. NLRP3 is formed by a PYD, NACHT, and LRR domain (Figure 1A). Many different studies have shown how NLRP3 can be activated by different triggers, suggesting the ubiquitous activation and importance of the NLRP3 inflammasome. NLRP3 can be activated by multiple infectious and endogenous ligands (Table 2), resulting in the most promiscuous of the inflammasomes. The different triggers of NLRP3 converges in some common cellular signaling as the decrease of the concentration of intracellular K+ (either by the activation of selective K+-conductance channels or plasma membrane permeabilization), production of reactive oxygen species (ROS), phagolysosome destabilization and the presence of oxidized mitochondrial DNA in the cytosol [35][36][37][38]. Therefore, NLRP3 can be considered as a sensor of the balanced intracellular environment, and some authors have suggested that NLRP3 is a sensor of HAMPs.
The canonical activation of NLRP3 in macrophages follow a two-step mechanism, where the first step is a priming of NLRP3 that is commonly achieved via Toll-like receptor (TLR) activation of nuclear factor kappa B (NF-κB). This priming results in the upregulation of NLRP3 gene expression and induction of different post-transcriptional modifications of NLRP3, such as de-ubiquitination or phosphorylation/dephosphorylation events among others [39]. This priming step could be also triggered by cytokines as tumor necrosis factor (TNF)-α or IL-1. The initial priming step also increases the expression of pro-IL-1β in macrophages. The second activation step of NLRP3 induces the homo-oligomerization of NLRP3 and is induced by a wide variety of different triggers (Table 2). NLRP3 oligomers recruit and oligomerize the adaptor protein ASC via PYD/PYD homotypic interactions and finally ASC recruit caspase-1 to promote its activation within the inflammasome.
Multiple other proteins can also bind to NLRP3 and are necessary for the activation of this inflammasome. Among them, never in mitosis gene a (NIMA)-related kinase 7 (NEK7), a member of the NIMA related kinases, has an important role in NLRP3 activation [40][41]. NEK7 acts downstream of the K+ efflux regulating the activation of NLRP3 by a direct binding to the NACHT and LRR domains [40]. However, NEK7 implication in NLRP3 activation can be bypassed by TAK1-dependent post-translational priming [42], suggesting that the activation mechanism of NLRP3 inflammasome can be tangled. Post-translational modifications of NLRP3 during the priming step, as different phosphorylation events in the PYD and NACHT domains [43][44][45][46], are important for a correct NLRP3 activation. In addition, interaction of NLRP3 with negatively charged lipids as the phosphatidylinositol-4-phophate on dispersed trans-Golgi network is necessary for its activation [47], confirming that the mechanism of NLRP3 activation is a complex pathway not fully understood yet. NLRP3 inflammasome can also be activated in a non-canonical manner after caspase-4/5 (in humans) or caspase-11 (in mouse) activation triggered by intracellular lipopolysaccharide (LPS) recognition. Caspase-4/5/11 activation cleaves GSDMD and N-terminal GSDMD pore formation induces K+ efflux and the non-canonical activation of the NLRP3 inflammasome [42].
NLRP3 has an important role in different pathologies, since the aberrant activation of this inflammasome is associated with several conditions, such as arthritis, gout, diabetes, Alzheimer’s disease or obesity [6]. Specific mutations in the NLRP3 gene have been associated with the development of a specific type of autoinflammatory manifestation termed cryopyrin-associated periodic syndromes (CAPS). CAPS individuals develop periodic skin rashes and fever [48], and the severity of these symptoms classify CAPS patients into: Neonatal-onset multisystem inflammatory disease (NOMID) as the most severe syndrome, Muckle-Wells syndrome (MWS) with intermediate symptoms, and the familial cold autoinflammatory syndrome (FCAS) as the milder form of CAPS triggered by low temperatures.
The fact that NLRP3 is involved in the initiation and progression of several human diseases lacking effective therapies has resulted in major advances on the understanding of NLRP3 activation mechanism and the development of several specific small molecules blocking NLRP3. However, we still do not fully understand the NLRP3 activation process, the mechanism of action of some NLRP3 blocking molecules, and their efficacy as novel drugs for humans.
2.3. NLRC4 Inflammasome
NLRC4 consists of a CARD, NACHT, and LRR domains (Figure 1A). The implication of ASC in this inflammasome is ambiguous because NLRC4 could directly promote caspase-1 activation, however, ASC seems necessary for IL-1β release [49], but not for pyroptosis [50] when NLRC4 is activated. NLRC4 does not directly bind triggering ligands as flagellin (Table 2), but there are proteins that belong to the NLR family of apoptosis inhibitory proteins (NAIPs) that recognize ligands and induce NLRC4 oligomerization. In mice, there are four different NAIPs, NAIP1 and NAIP2 recognize the bacterial needle and inner rod proteins, and NAIP5 and NAIP6 bind to flagellin [51][52][53][54]. Only one NAIP is present in humans, and this NAIP is able to sense the different NLRC4 activators [52][53][54]. The phosphorylation of Ser533 is important for NLRC4 activation after Salmonella infection, because it probably drives a conformational change in NLRC4 important for its activation [55]. Therefore, similarly to NLRP3, post-translational modifications in NLRC4 are needed for its activation.
Gain-of-function mutations in NLCR4 gene have been associated with autoinflammatory diseases [17][56][57][58], with symptoms overlapping CAPS and in the most severe syndromes auto-active NLRC4 mutations associate with the macrophage activating syndrome (MAS). Differently from CAPS, the macrophage activating syndrome is mainly mediated by IL-18 and treatment with recombinant IL-18 binding protein has been successfully used in patients [56]. The design of specific inhibitors blocking NLRC4 is an important area of development, however, no small molecules blocking NLRC4 have been described so far.
2.4. NLRP6 Inflammasome
NLRP6 is an inflammasome with important roles as immunosensor. Its structure consists of a PYD, NACHT, and LRR domains (Figure 1A), but even sharing a 43.8% of similarity with NLRP3, NLRP6 cannot be activated by the same stimuli that NLRP3 senses (Table 2). The mechanism of activation and action of NLRP6 has not been well described to date, however it has been reported to form an inflammasome by interacting with ASC, but this results in caspase-11 activation instead of caspase-1 [59]. The PYD domain from NLRP6 is able to self-assemble in a filament manner that can bind ASC [60]. Most of the studies about the NLRP6 inflammasome have been focused in cells of the intestine, where it is highly expressed, and one of the main physiological roles of NLRP6 is to maintain the intestinal mucosal homeostasis in an optimal state [61][62]. Other physiological functions of NLRP6 have been described, including the inhibition in carcinogenesis and promoting neuroinflammation [63][64]. To date, no pathological mutations have been associated with NLRP6, and no specific inhibitors have been designed for this inflammasome.
3. Non-NLR Inflammasomes
3.1. AIM2 Inflammasome
AIM2 is the only member of the AIM2-like receptor (ALR) family that has been described to form an inflammasome. AIM2 presents a PYD and a HIN domain (Figure 1B), this last one is a domain that directly binds dsDNA (Table 2), dsDNA being a potent activator of this inflammasome [65]. The binding of AIM2 to dsDNA is independent of the origin or sequence of the DNA, although a length larger than 80 bp is needed to obtain a robust AIM2 activation [4]. The fact that AIM2 does not have a CARD domain makes the requirement of ASC necessary for the activation of caspase-1 within this inflammasome [66]. Other protein partners have been described to be required for AIM2 activation, and for example during Francisella novocida infection, the presence of guanylate binding proteins (GBP), specifically GBP2 and GBP5, are necessary to activate AIM2 [67].
Physiologically, AIM2 is considered as a therapeutic target for autoimmune disorders, specifically in psoriasis and systemic lupus erythematosus [68][69] where endogenous dsDNA is a trigger of these conditions. Additionally, an interesting role for AIM2 in cancer therapeutics has been described, where AIM2 mediates the caspase-1 dependent death of intestinal epithelial and bone marrow cells in response to dsDNA breaks produced by radiotherapy and chemotherapeutic agents, without affecting the anti-cancer properties of these drugs. This suggests that AIM2 could be modulated to decrease or limit the effects of chemotherapy [70].
3.2. Pyrin Inflammasome
Pyrin protein is formed by a PYD, a central coiled BBOX domain and a C-terminal B30.2/SPRY domain (SPia and the Ryanodine Receptor domain) (Figure 1B), this last domain is not present in the mouse Pyrin orthologous proteins. Unlike other inflammasomes, Pyrin is activated by pathogen-mediated modifications of host proteins (Table 2) and once is activated, ASC binds to the PYD domain to form an inflammasome, which activates caspase-1, IL-1β release, and GSDMD-dependent pyroptosis [71][72]. The protein 14-3-3 is important for Pyrin activation, as its binding to the phosphorylated Ser242 residue in Pyrin keeps this inflammasome inhibited. Pyrin dephosphorylation induced by small GTPases inhibition releases Pyrin from 14-3-3 and promotes Pyrin interaction with ASC [73]. Different bacterial toxins, as Clostridium difficile toxin B (TcdB) are potent inhibitors of GTPases, and therefore strong activators of the Pyrin inflammasome [74].
Pyrin inflammasome is important for the host response during bacterial infections and the deficiency of Pyrin increase bacterial load in macrophages [75]. Mutations in Pyrin have been associated with autoinflammatory human diseases, in particular to Familial Mediterranean fever [76], but also to myalgia, serositis, amyloidosis, and neutrophilic dermatosis [77]. Colchicine is the first line of drug used to treat familial Mediterranean fever, and anti-IL-1 therapy is used in case the patient is resistant to colchicine. However, any of these compounds directly block Pyrin inflammasome, therefore there is an urgent need to find specific Pyrin blockers.
References
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426.
- Hara, H.; Seregin, S.S.; Yang, D.; Fukase, K.; Chamaillard, M.; Alnemri, E.S.; Inohara, N.; Chen, G.Y.; Núñez, G. The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell 2018, 175, 1651–1664.e14.
- Vanaja, S.K.; Rathinam, V.A.K.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015.
- Rathinam, V.A.K.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 2012, 13, 333–342.
- De Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287.
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687.
- De Alba, E. Structure, interactions and self-assembly of ASC-dependent inflammasomes. Arch. Biochem. Biophys. 2019, 670, 15–31.
- Franklin, B.S.; Latz, E.; Schmidt, F.I. The intra- and extracellular functions of ASC specks. Immunol. Rev. 2018, 281, 74–87.
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840.
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665.
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116.
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673.
- Jiang, D.; Chen, S.; Sun, R.; Zhang, X.; Wang, D. The NLRP3 inflammasome: Role in metabolic disorders and regulation by metabolic pathways. Cancer Lett. 2018, 419, 8–19.
- Shen, H.H.; Yang, Y.X.; Meng, X.; Luo, X.Y.; Li, X.M.; Shuai, Z.W.; Ye, D.Q.; Pan, H.F. NLRP3: A promising therapeutic target for autoimmune diseases. Autoimmun. Rev. 2018, 17, 694–702.
- Booshehri, L.M.; Hoffman, H.M. CME REVIEW CAPS and NLRP3. J. Clin. Immunol. 2019, 39, 277–286.
- Moghaddas, F.; Zeng, P.; Zhang, Y.; Schützle, H.; Brenner, S.; Hofmann, S.R.; Berner, R.; Zhao, Y.; Lu, B.; Chen, X.; et al. Autoinflammatory mutation in NLRC4 reveals a leucine-rich repeat (LRR)–LRR oligomerization interface. J. Allergy Clin. Immunol. 2018, 142, 1956–1967.e6.
- Malhotra, S.; Costa, C.; Eixarch, H.; Keller, C.W.; Amman, L.; Martínez-Banaclocha, H.; Midaglia, L.; Sarró, E.; Machín-Díaz, I.; Villar, L.M.; et al. NLRP3 inflammasome as prognostic factor and therapeutic target in primary progressive multiple sclerosis patients. Brain 2020, 143, 1414–1430.
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2020, 1–12.
- Knorr, J.; Wree, A.; Tacke, F.; Feldstein, A.E. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin. Liver Dis. 2020, 40, 298–306.
- Amores-Iniesta, J.; Barberà-Cremades, M.; Martínez, C.M.; Pons, J.A.; Revilla-Nuin, B.; Martínez-Alarcón, L.; Di Virgilio, F.; Parrilla, P.; Baroja-Mazo, A.; Pelegrín, P. Extracellular ATP Activates the NLRP3 Inflammasome and Is an Early Danger Signal of Skin Allograft Rejection. Cell Rep. 2017, 21, 3414–3426.
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489.
- Ting, J.P.Y.; Lovering, R.C.; Alnemri, E.S.P.D.; Bertin, J.; Boss, J.M.; Davis, B.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A.; et al. The NLR gene family: An official nomenclature. Immunity 2008, 28, 285–287.
- Eldeeb, M.A.; Fahlman, R.P.; Esmaili, M.; Fon, E.A. When Degradation Elicits the Alarm: N-Terminal Degradation of NLRP1B Unleashes Its Inflammasome Activity. Mol. Cell 2019, 74, 637–639.
- Boyden, E.D.; Dietrich, W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006, 38, 240–244.
- Chui, A.J.; Okondo, M.C.; Rao, S.D.; Gai, K.; Griswold, A.R.; Johnson, D.C.; Ball, D.P.; Taabazuing, C.Y.; Orth, E.L.; Vittimberga, B.A.; et al. N-terminal degradation activates the NLRP1B inflammasome. Science 2019, 364, 82–85.
- Sandstrom, A.; Mitchell, P.S.; Goers, L.; Mu, E.W.; Lesser, C.F.; Vance, R.E. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 2019, 364.
- Frew, B.C.; Joag, V.R.; Mogridge, J. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog. 2012, 8.
- Gorfu, G.; Cirelli, K.M.; Melo, M.B.; Mayer-Barber, K.; Crown, D.; Koller, B.H.; Masters, S.; Sher, A.; Leppla, S.H.; Moayeri, M.; et al. Dual Role for Inflammasome Sensors NLRP1 and NLRP3 in Murine Resistance to Toxoplasma gondii. MBio 2014, 5.
- Witola, W.H.; Mui, E.; Hargrave, A.; Liu, S.; Hypolite, M.; Montpetit, A.; Cavailles, P.; Bisanz, C.; Cesbron-Delauw, M.F.; Fournié, G.J.; et al. NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect. Immun. 2011, 79, 756–766.
- Yu, C.H.; Moecking, J.; Geyer, M.; Masters, S.L. Mechanisms of NLRP1-Mediated Autoinflammatory Disease in Humans and Mice. J. Mol. Biol. 2018, 430, 142–152.
- Jin, Y.; Mailloux, C.M.; Gowan, K.; Riccardi, S.L.; Laberge, G.; Bennett, D.C.; Fain, P.R.; Spritz, R.A. NALP1 in vitiligo-associated multiple autoimmune disease. N. Engl. J. Med. 2007, 356, 1216–1225.
- Zurawek, M.; Fichna, M.; Januszkiewicz-Lewandowska, D.; Gryczyńska, M.; Fichna, P.; Nowak, J. A coding variant in NLRP1 is associated with autoimmune Addison’s disease. Hum. Immunol. 2010, 71, 530–534.
- Zhong, F.L.; Mamaï, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverényi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 2016, 167, 187–202.e17.
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153.
- Zhou, R.; Yazdi, A.S.; Menu, P. A role for mitochondria in NLRP3 inflammasome activation. Nature 2010, 1–6.
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856.
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414.
- Baker, P.J.; De Nardo, D.; Moghaddas, F.; Tran, L.S.; Bachem, A.; Nguyen, T.; Hayman, T.; Tye, H.; Vince, J.E.; Bedoui, S.; et al. Posttranslational modification as a critical determinant of cytoplasmic innate immune recognition. Physiol. Rev. 2017, 97, 1165–1209.
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Núñez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357.
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258.
- Schmacke, N.A.; Gaidt, M.M.; Szymanska, I.; O’Duill, F.; Stafford, C.A.; Chauhan, D.; Fröhlich, A.L.; Nagl, D.; Pinci, F.; Schmid-Burgk, J.L.; et al. Priming enables a NEK7-independent route of NLRP3 activation. bioRxiv 2019, 799320.
- Stutz, A.; Kolbe, C.C.; Stahl, R.; Horvath, G.L.; Franklin, B.S.; Van Ray, O.; Brinkschulte, R.; Geyer, M.; Meissner, F.; Latz, E. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 2017, 214, 1725–1736.
- Song, N.; Liu, Z.S.; Xue, W.; Bai, Z.F.; Wang, Q.Y.; Dai, J.; Liu, X.; Huang, Y.J.; Cai, H.; Zhan, X.Y.; et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol. Cell 2017, 68, 185–197.e6.
- Zhang, Z.; Meszaros, G.; He, W.T.; Xu, Y.; de Fatima Magliarelli, H.; Mailly, L.; Mihlan, M.; Liu, Y.; Gámez, M.P.; Goginashvili, A.; et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med. 2017, 214, 2671–2693.
- Spalinger, M.R.; Kasper, S.; Gottier, C.; Lang, S.; Atrott, K.; Vavricka, S.R.; Scharl, S.; Gutte, P.M.; Grütter, M.G.; Beer, H.D.; et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J. Clin. Investig. 2016, 126, 1783–1800.
- Chen, J.; Chen, Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564, 71–76.
- Hoffman, H.M.; Mueller, J.L.; Broide, D.H.; Wanderer, A.A.; Kolodner, R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 2001, 29, 301–305.
- Van Opdenbosch, N.; Gurung, P.; Vande Walle, L.; Fossoul, A.; Kanneganti, T.D.; Lamkanfi, M. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat. Commun. 2014, 5, 1–14.
- Broz, P.; Von Moltke, J.; Jones, J.W.; Vance, R.E.; Monack, D.M. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 2010, 8, 471–483.
- Kofoed, E.M.; Vance, R.E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 2012, 477, 592–595.
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.N.; Lu, Q.; Xu, H.; Liu, L.; Shao, F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011, 477, 596–602.
- Rayamajhi, M.; Zak, D.E.; Chavarria-Smith, J.; Vance, R.E.; Miao, E.A. Cutting edge: Mouse NAIP1 detects the type III secretion system needle protein. J. Immunol. 2013, 191, 3986–3989.
- Yang, J.; Zhao, Y.; Shi, J.; Shao, F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc. Natl. Acad. Sci. USA 2013, 110, 14408–14413.
- Qu, Y.; Misaghi, S.; Izrael-Tomasevic, A.; Newton, K.; Gilmour, L.L.; Lamkanfi, M.; Louie, S.; Kayagaki, N.; Liu, J.; Kömüves, L.; et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 2012, 490, 539–542.
- Canna, S.W.; de Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.M.; Sanchez, G.A.M.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146.
- Kitamura, A.; Sasaki, Y.; Abe, T.; Kano, H.; Yasutomo, K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J. Exp. Med. 2014, 211, 2385–2396.
- Romberg, N.; Al Moussawi, K.; Nelson-Williams, C.; Stiegler, A.L.; Loring, E.; Choi, M.; Overton, J.; Meffre, E.; Khokha, M.K.; Huttner, A.J.; et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 2014, 46, 1135–1139.
- Grenier, J.M.; Wang, L.; Manji, G.A.; Huang, W.J.; Al-Garawi, A.; Kelly, R.; Carlson, A.; Merriam, S.; Lora, J.M.; Briskin, M.; et al. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-κB and caspase-1. FEBS Lett. 2002, 530, 73–78.
- Shen, C.; Lu, A.; Xie, W.J.; Ruan, J.; Negro, R.; Egelman, E.H.; Fu, T.M.; Wu, H. Molecular mechanism for NLRP6 inflammasome assembly and activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2052–2057.
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 156, 1045–1059.
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757.
- Chen, G.Y.; Liu, M.; Wang, F.; Bertin, J.; Núñez, G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 2011, 186, 7187–7194.
- Ydens, E.; Demon, D.; Lornet, G.; De Winter, V.; Timmerman, V.; Lamkanfi, M.; Janssens, S. Nlrp6 promotes recovery after peripheral nerve injury independently of inflammasomes. J. Neuroinflammation 2015, 12, 1–14.
- Roberts, T.L.; Idris, A.; Dunn, J.A.; Kelly, G.M.; Burnton, C.M.; Hodgson, S.; Hardy, L.L.; Garceau, V.; Sweet, M.J.; Ross, I.L.; et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009, 323, 1057–1060.
- Schattgen, S.A.; Fitzgerald, K.A. The PYHIN protein family as mediators of host defenses. Immunol. Rev. 2011, 243, 109–118.
- Meunier, E.; Wallet, P.; Dreier, R.F.; Costanzo, S.; Anton, L.; Rühl, S.; Dussurgey, S.; Dick, M.S.; Kistner, A.; Rigard, M.; et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat. Immunol. 2015, 16, 476–484.
- Dombrowski, Y.; Peric, M.; Koglin, S.; Kammerbauer, C.; Göß, C.; Anz, D.; Simanski, M.; Gläser, R.; Harder, J.; Hornung, V.; et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci. Transl. Med. 2011, 3, 1–16.
- Choubey, D. Interferon-inducible Ifi200-family genes as modifiers of lupus susceptibility. Immunol. Lett. 2012, 147, 10–17.
- Hu, B.; Jin, C.; Li, H.-B.; Tong, J.; Ouyang, X.; Cetinbas, N.M.; Zhu, S.; Strowig, T.; Lam, F.C.; Zhao, C.; et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 2016, 354, 765–768.
- Chae, J.J.; Komarow, H.D.; Cheng, J.; Wood, G.; Raben, N.; Liu, P.P.; Kastner, D.L. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol. Cell 2003, 11, 591–604.
- Richards, N.; Schaner, P.; Diaz, A.; Stuckey, J.; Shelden, E.; Wadhwa, A.; Gumucio, D.L. Interaction between Pyrin and the Apoptotic Speck Protein (ASC) Modulates ASC-induced Apoptosis. J. Biol. Chem. 2001, 276, 39320–39329.
- Masters, S.L.; Lagou, V.; Jéru, I.; Baker, P.J.; Van Eyck, L.; Parry, D.A.; Lawless, D.; De Nardo, D.; Garcia-Perez, J.E.; Dagley, L.F.; et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci. Transl. Med. 2016, 8, 332ra45.
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.N.; Peng, X.; Xi, J.J.; Chen, S.; et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014, 513, 237–241.
- Broecker, F.; Andrae, K.; Moelling, K. Premature activation of the HIV RNase H drives the virus into suicide: A novel microbicide? AIDS Res. Hum. Retrovir. 2012, 28, 1397–1403.
- Chae, J.J.; Cho, Y.H.; Lee, G.S.; Cheng, J.; Liu, P.P.; Feigenbaum, L.; Katz, S.I.; Kastner, D.L. Gain-of-Function Pyrin Mutations Induce NLRP3 Protein-Independent Interleukin-1β Activation and Severe Autoinflammation in Mice. Immunity 2011, 34, 755–768.
- De Torre-Minguela, C.; del Castillo, P.M.; Pelegrín, P. The NLRP3 and pyrin inflammasomes: Implications in the pathophysiology of autoinflammatory diseases. Front. Immunol. 2017, 8.



