 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dragan Copic | + 1177 word(s) | 1177 | 2021-03-16 04:38:27 | | | |
| 2 | Vicky Zhou | Meta information modification | 1177 | 2021-03-23 10:28:43 | | |
Video Upload Options
Ichthyoses comprise a broad spectrum of keratinization disorders due to hereditary defects of cornification. Until now, mutations in more than 50 genes, mostly coding for structural proteins involved in epidermal barrier formation, have been identified as causes for different types of these keratinization disorders. However, due to the high heterogeneity and difficulties in the establishment of valid experimental models, research in this field remains challenging and translation of novel findings to clinical practice is difficult.
1. Introduction
The spectrum of hereditary disorders of cornification (DOC) consists of a variety of rare genodermatoses, such as ichthyoses and palmoplantar keratodermas [1][2]. For many of these diseases, the underlying pathological causes have been identified. Most frequently, these disorders develop due to monogenic alterations in genes important for the establishment or maintenance of a functional epidermal barrier. These genes are essential for a variety of epidermal processes, including epidermal lipid metabolism, the establishment of intercellular junctions and keratinocyte differentiation [3][4]. Patients with mutations in these genes develop symptoms, such as epidermal thickening, scaling, and increased susceptibility for concomitant inflammation of the skin [2][5].
Ichthyoses represent a large and heterogeneous group of hereditary DOC [6]. They are classified by inheritance patterns, their underlying pathomechanisms, and clinical features, and can be divided into syndromic and non-syndromic forms [7]. Ichthyosis vulgaris (IV) is the most common ichthyosis. It develops due to mutations in the filaggrin gene (FLG, OMIM #146700) and manifests with dry and scaly skin without overt signs of inflammation [8][9][10]. Histologically, IV epidermis is characterized by massive hyperkeratosis and a reduction or lack of keratohyalin granules [11]. In contrast to IV, autosomal recessive congenital ichthyoses (ARCI) are a group of rare skin disorders and include, amongst others, lamellar ichthyosis (LI) and harlequin ichthyosis (HI) [12]. Depending on the underlying mutation, manifesting symptoms are strongly variable [12]. Affected patients are characterized by patchy or generalized scaling that evolves into large scaly plaques and erythema. Netherton syndrome (NS), a syndromic form of ichthyosis, is caused by loss of function mutation in the serine protease inhibitor of Kazal type 5-gene (SPINK5, OMIM #256500) [13]. Functional loss of the SPINK5-encoded serine protease inhibitor lympho-epithelial Kazal type related inhibitor (LEKTI) leads to a compromised inhibition of kallikreins. The balance between protease activity and inhibition is shifted towards excessive proteolytic activity, leading to premature degradation of corneodesmosomes, impaired skin barrier integrity and skin inflammation [13].
The heterogeneous group of peeling skin syndromes (PSS) are autosomal recessive diseases that can be divided into localized and generalized forms [14]. Mutations of transglutaminase 5 (TGM5, OMIM #609796) and cystatin A (CSTA, OMIM #607936) have been identified in patients affected with localized PSS [15]. Within the generalized PSS, a further distinction can be made between a generalized non-inflammatory PSS (Type A PSS) and a generalized PSS with inflammation (Type B PSS) [15]. While Type A PSS is associated with loss-of-function mutations in the peptidase inhibitor clade B member 8 (SERPINB8, OMIM #617115) and filaggrin 2 (FLG2, OMIM #618084) gene, Type B PSS manifests after loss of function mutations in the corneodesmosin (CDSN, OMIM #270300) gene [16].
Hereditary palmoplantar keratodermas (PPKs) represent another heterogenic disease cluster [17]. The morphological characteristics of PPKs are reflected by hyperkeratosis and skin thickening on palms and soles [18]. Clinically, diffuse and circumscribed or punctiform keratosis can be distinguished. The spectrum of genes involved in the many different forms of PPKs is very broad [19]. Mutations of genes encoding the cornified envelope protein loricrin, several keratins, connexins, and other proteins have been identified in these skin disorders [17][19].
A common trait of most of these diseases is their early manifestation during infancy or young adulthood and the considerable physical and psychological burden for the affected patients. Therapeutic strategies mainly aim to improve the quality of life of the patients by relieving symptoms and by attempting to restore the functionality of the skin [20]. Approved treatment options include frequent use of emollients for moisturization, keratolytic compounds for the hyperkeratotic areas, and retinoids to promote normal keratinocyte differentiation [20]. In addition, prevention of bacterial and fungal skin infections is an important consideration when treating patients suffering from hereditary DOC [20]. As currently no cure for hereditary DOCs exists, validated disease models are highly desirable to extend the current understanding of the underlying pathomechanisms and to foster the development of novel and effective treatment options.
2. Models to Study Hereditary DOC
Due to usually low patient numbers, studies on patients affected with hereditary DOCs are strongly limited and patient-derived samples are hardly available. Therefore, the establishment of valid disease models has been indispensable to study disease-related mechanisms and to develop new treatment options. Several experimental approaches have already been shown to significantly contribute to a better understanding of DOCs, including in vivo animal models, in vitro skin models, and in silico studies (Figure 1).
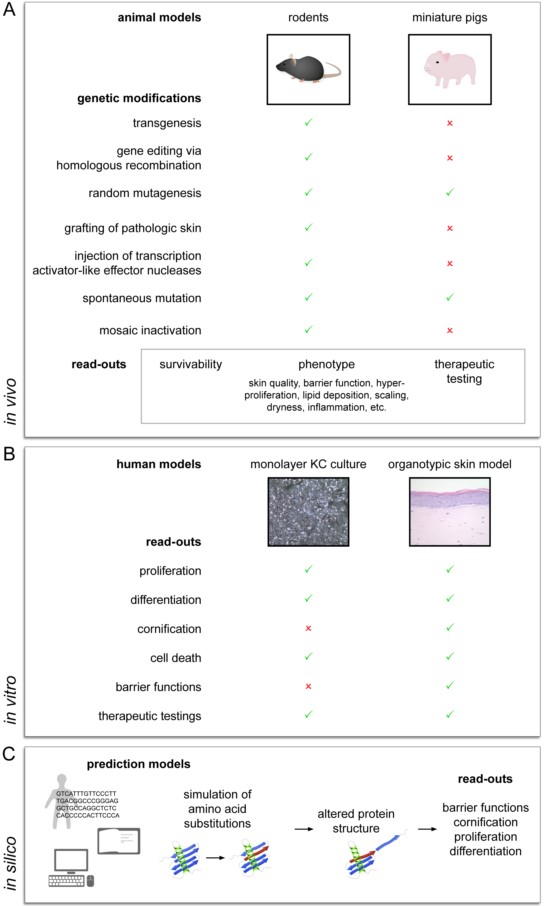
3. Conclusions
Hereditary DOCs bear a great burden to affected individuals, and due to their low frequency, studies in humans are scarce. Therefore, valid experimental models are needed to better understand the underlying pathomechanisms and develop new therapeutic strategies for patients. In vivo animal approaches offer valuable insights into the complex interactions of the skin with other organ systems, however, there are considerable limitations to their use for the study of hereditary DOCs. Especially in settings involving small animal models, the early onset of postnatal death makes the study of long-term complications difficult. Additionally, interspecies differences in structural composition and physiological function of the epidermis further complicate the translation of findings of experimental in vivo models to clinical settings. Following the European ban on animal testing, the 3R principles (Replacement, Reduction, Refinement) for research involving animal models were implemented. Ever since, non-animal-based models have moved to the frontline and gradually replaced animal-based approaches. Three-dimensional human organotypic skin models are well suited to investigate biological processes involved in hereditary DOCs. By using gene silencing methods, several of the described pathologies have already been reproduced in human organotypic skin models. The use of human material represents a major advantage of this method. Another big advantage is its potential to easily and rapidly screen for potential candidate genes that so far may not have been associated with hereditary DOCs. A disadvantage, however, is that interactions with or the influence of other cell types or organs cannot be studied in these in vitro skin models. The use of in silico methods to study DOCs is still at the beginning. However, it is very likely that new bioinformatics tools and in silico predictions will strongly impact the research on DOCs in the near future.
Although the establishment of models for DOCs remains challenging, constant progress in developing new methods and new models has led to a better understanding of these pathologies.
References
- Ammirati, C.T.; Mallory, S.B. The major inherited disorders of cornification: New advances in pathogenesis. Dermatol. Clin. 1998, 16, 497–508.
- Mathes, E.F.; Spring, S.; Friedland, R. PAS. Hereditary Disorders of Cornification. In Therapy in Pediatric Dermatology; Teng, J., Marqueling, A., Benjamin, L., Eds.; Springer: Cham, Switzerland, 2017.
- Van Smeden, J.; Janssens, M.; Gooris, G.S.; Bouwstra, J.A. The important role of stratum corneum lipids for the cutaneous barrier function. Biochim. Biophys. Acta–Mol. Cell Biol. Lipids 2014, 1841, 295–313.
- Candi, E.; Schmidt, R.; Melino, G. The cornified envelope: A model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 2005, 6, 328–340.
- Bouwstra, J.A.; Ponec, M. The skin barrier in healthy and diseased state. Biochim. Biophys. Acta-Biomembr. 2006, 1758, 2080–2095.
- Schmuth, M.; Martinz, V.; Janecke, A.R.; Fauth, C.; Schossig, A.; Zschocke, J.; Gruber, R. Inherited ichthyoses/generalized Mendelian disorders of cornification. Eur. J. Hum. Genet. 2013, 21, 123–133.
- Oji, V.; Tadini, G.; Akiyama, M.; Blanchet Bardon, C.; Bodemer, C.; Bourrat, E.; Coudiere, P.; DiGiovanna, J.; Elias, P.; Fischer, J.; et al. Revised nomenclature and classification of inherited ichthyoses: Results of the First Ichthyosis Consensus Conference in Sorze 2009. J. Am. Acad. Dermatol. 2010, 63, 607–641.
- Takeichi, T.; Akiyama, M. Inherited ichthyosis: Non-syndromic forms. J. Dermatol. 2016, 43, 242–251.
- McLean, W.H.I. Filaggrin failure—From ichthyosis vulgaris to atopic eczema and beyond. Br. J. Dermatol. 2016, 175, 4–7.
- Thyssen, J.P.; Godoy-Gijon, E.; Elias, P.M. Ichthyosis vulgaris: The filaggrin mutation disease. Br. J. Dermatol. 2013, 168, 1155–1166.
- Rodríguez-Pazos, L.; Ginarte, M.; Vega, A.; Toribio, J. Autosomal recessive congenital ichthyosis. Actas Dermosifiliogr. 2013, 104, 270–284.
- Hovnanian, A. Netherton syndrome: Skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res. 2013, 351, 289–300.
- Oji, V.; Metze, D.; Traupe, H. Inherited Disorders of Cornification. In Rook’s Textbook of Dermatology, 9th ed.; Griffiths, C.E.M., Barker, J., Bleiker, T., Chalmers, R., Creamer, D., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2016.
- Has, C. Peeling Skin Disorders: A Paradigm for Skin Desquamation. J. Investig. Dermatol. 2018, 138, 1689–1691.
- Samuelov, L.; Sprecher, E. Peeling off the genetics of atopic dermatitis-like congenital disorders. J. Allergy Clin. Immunol. 2014, 134, 808–815.
- Has, C.; Technau-Hafsi, K. Palmoplantar keratodermas: Clinical and genetic aspects. J. Der Dtsch. Dermatologischen Gesellschaft 2016, 14, 123–140.
- Thomas, L.J.; Freeman, A.; O’Toole, E.A.; McGrath, J.A.; Perrett, C.M. Inherited palmoplantar keratodermas: The heart of the matter. Clin. Exp. Dermatol. 2018, 43, 228–230.
- Sakiyama, T.; Kubo, A. Hereditary palmoplantar keratoderma “clinical and genetic differential diagnosis”. J. Dermatol. 2016, 43, 264–274.
- Oji, V.; Preil, M.L.; Kleinow, B.; Wehr, G.; Fischer, J.; Hennies, H.C.; Hausser, I.; Breitkreuz, D.; Aufenvenne, K.; Stieler, K.; et al. S1 guidelines for the diagnosis and treatment of ichthyoses—Update. J. Ger. Soc. Dermatol. 2017, 15, 1053–1065.
- Avci, P.; Sadasivam, M.; Gupta, A.; De Melo, W.C.; Huang, Y.Y.; Yin, R.; Chandran, R.; Kumar, R.; Otufowora, A.; Nyame, T.; et al. Animal models of skin disease for drug discovery. Expert Opin. Drug Discov. 2013, 8, 331–355.

