 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marco Scarselli | + 4273 word(s) | 4273 | 2021-03-15 09:51:20 | | | |
| 2 | Vivi Li | + 72 word(s) | 4345 | 2021-03-23 08:40:54 | | |
Video Upload Options
Atypical antipsychotics (AAPs) are commonly prescribed medications to treat schizophrenia, bipolar disorders and other psychotic disorders. However, they might cause metabolic syndrome (MetS) in terms of weight gain, dyslipidemia, type 2 diabetes (T2D), and high blood pressure, which are responsible for reduced life expectancy and poor adherence. Importantly, there is clear evidence that early metabolic disturbances can precede weight gain, even if the latter still remains the hallmark of AAPs use. In fact, AAPs interfere profoundly with glucose and lipid homeostasis acting mostly on hypothalamus, liver, pancreatic β-cells, adipose tissue, and skeletal muscle. Their actions on hypothalamic centers via dopamine, serotonin, acetylcholine, and histamine receptors affect neuropeptides and 5′AMP-activated protein kinase (AMPK) activity, thus producing a supraphysiological sympathetic outflow augmenting levels of glucagon and hepatic glucose production. In addition, altered insulin secretion, dyslipidemia, fat deposition in the liver and adipose tissues, and insulin resistance become aggravating factors for MetS. In clinical practice, among AAPs, olanzapine and clozapine are associated with the highest risk of MetS, whereas quetiapine, risperidone, asenapine and amisulpride cause moderate alterations. The new AAPs such as ziprasidone, lurasidone and the partial agonist aripiprazole seem more tolerable on the metabolic profile. However, these aspects must be considered together with the differences among AAPs in terms of their efficacy, where clozapine still remains the most effective. Intriguingly, there seems to be a correlation between AAP’s higher clinical efficacy and increase risk of metabolic alterations. Finally, a multidisciplinary approach combining psychoeducation and therapeutic drug monitoring (TDM) is proposed as a first-line strategy to avoid the MetS. In addition, pharmacological treatments are discussed as well.
1. Introduction
Antipsychotic (AP) medications are associated with relevant metabolic side effects, where the so-called metabolic syndrome (MetS) can be responsible for poor adherence, sub-optimal and discontinuation drug use, resulting in relapse and poor clinical outcome. In fact, about 20–50% of patients suffering from schizophrenia or other psychotic disorders, in the long-term, may suspend drug usage with a detrimental effect for their prognosis [1].
In addition, MetS, which has an occurrence of about 40% in chronic schizophrenic patients, has a relevant impact on their general health conditions [1][2]. MetS is defined by the presence of metabolic abnormalities, such as large waist circumference, dyslipidemia, fasting hyperglycemia and elevated blood pressure. Schizophrenic patients have higher morbidity and mortality compared to the general population, where cardiovascular problems are the main cause of this decease and the estimated life expectancy drops down by 10–20 years [3]. However, recent data have confirmed that long term use of atypical antipsychotics (AAPs) in the schizophrenic population is associated with decreased hospitalization and mortality compared to untreated patients, especially with the use of clozapine [4][5].
The relationship between MetS and schizophrenia is complex and multifactorial, where the use of APs has a major role. Besides drugs side effects, other factors such as unhealthy lifestyle, reduced physical activity, smoking, improper diet and genetic predisposition also contribute to metabolic disturbances [6].
APs are generally divided into typical antipsychotics (TAPs) or first-generation APs and atypical antipsychotics (AAPs) or second-generation APs, based on the evidence that AAPs rarely induce motor side effects. However, this distinction has been questioned by many, so a new classification among AAPs was recently proposed by introducing the concept of spectrum of atypia that begins with risperidone (the least atypical) and ends with clozapine (the most atypical), which is still the gold standard for the treatment-resistant schizophrenia [7].
Unfortunately, AAPs higher efficacy on cognition and negative symptoms of schizophrenia is outweighed by the frequent occurrence of MetS and weight gain, thereby limiting their use in clinical practice and compelling clinicians for constant monitoring of patient conditions. It is quite surprising that these relevant side effects, in particular for AAPs, have been underestimated in the past, but now finally clinicians are addressing these concerns while treating psychotic disorders.
Different meta-analysis have shown that the use of AAPs is associated with the highest risk of MetS compared with TAPs, with clozapine and olanzapine being the worse [8]. Importantly, type 2 diabetes (T2D) is not strictly correlated with adiposity. In fact, 25% of patients develop hyperglycemia without gaining weight and this metabolic alteration can happen in the early period of the treatments and precede weight gain [9]. On the other hand, it is important to underline, in the long-term, how weight gain induced by AAPs undoubtedly represents an aggravating factor in the whole metabolic regulation.
The molecular and cellular mechanisms responsible for these metabolic changes are complex and involve practically all the organs relevant for metabolism, including the central and peripheral nervous system.
Indeed, AAPs are drugs targeting many receptors and other proteins, and significantly affect the activities of many hormones and neuromodulators. Different affinities of AAPs on dopamine, serotonin, muscarinic, adrenergic, histamine receptors and other molecular targets (e.g., AMPK) are responsible of their diverse clinical profiles. These receptor targets are well expressed in the hypothalamic centers, pancreas, liver, adipose tissue and skeletal muscle where they modulate glucose and lipid homeostasis in the whole body [10].
With these premises, this review aims to analyze the occurrence of MetS during the use of AAPs by trying to understand the responsible mechanisms in order to identify new potential targets to avoid and/or reduce this undesired effect.
A multidisciplinary approach combining psychoeducation and therapeutic drug monitoring (TDM) is proposed as a first-line strategy. In addition, pharmacological treatments are discussed as well.
2. A New Classification for AAPs: The Spectrum of Atypia
Since the discovery of clozapine in the 70’s, the concept of AAPs or second-generation APs was introduced, referring to a new class of medications that were better tolerable, especially in terms of motor-related side effects. However, in the following years, it emerged that the benefits of this new class of drugs were somehow overshadowed due to induced metabolic side effects such as obesity and diabetes. In addition, clinical evidence underlined the diversity within the class of AAPs in terms of efficacy and motor and endocrine-related side effects, highlighting how each AAP was unique and therefore the differences among TAPs and AAPs were questionable [11].
To reconcile the concept of atypicality and diversity among the class of AAPs, a new classification for AAPs was recently proposed by introducing the notion of spectrum of atypia, that ranges from risperidone, the least atypical, to clozapine, the most atypical, while all the other AAPs fall within the extremes of this spectrum (Figure 1). Notably, risperidone and amisulpride can lose their atypicality at higher doses [7]. With this clarification, the concept of atypia is still intact in its essence referring to a category of APs, which demonstrate reduced motor problems, reduced hyperprolactinemia, and reduced worsening of apathy and anhedonia along with a better improvement of negative and cognitive symptoms of schizophrenia. The most effective AAP is still clozapine, which has unique clinical properties to treat drug-refractory schizophrenia, psychoses associated with Parkinson’s disease (PD) and tardive dyskinesia. Unfortunately, these advantages have to be balanced with the increased risk of the AAPs-induced MetS, which is seen less with TAPs [12].

Figure 1. The concept of spectrum of atypia was recently introduced to classify atypical antipsychotics (AAPs). They can be divided in three categories, where risperidone is least atypical (Level I) and clozapine is most atypical (Level III), while all others fall within these two extremes of the spectrum (Level II). The molecular targets shown on the right add up, beginning with the D2 and 5-HT2A,C receptors that are common targets for all AAPs, extending to additional mechanisms such as M1 positive allosterism and GlyT (glycine transporter) activity that seem specific to clozapine. Other targets, such as H1 and α2 receptors and BDNF, are relevant to both Level II and III of atypia [7].
In fact, in clinical practice, many doctors make decisions for a tailored therapy according to the patient’s characteristics, drug efficacy and risks of drug-related side effects. As a consequence, the choice among the different APs is often made by trying to avoid the risk of motor-related side effects or metabolic abnormalities.
Regarding the mechanism of action, AAPs are weak D2 receptor blockers and they act beyond D2 antagonism, involving other receptor targets (e.g., serotonin (5-HT) receptors) [13][14][15][16]. The ratio of 5-HT2A/D2 and 5-HT2C/D2 receptor affinity together with a rapid dissociation constant (Koff) from the D2 receptor are two important factors that distinguish AAPs in terms of efficacy and side effects [17][18]. In addition, other molecular targets characterize the receptor profile of the ideal AAP, and among them, 5-HT1 partial agonism, H1 antagonism, α2 antagonism, muscarinic antagonism and positive allosterism, brain-derived neurotrophic factor (BDNF) production, and glycine transporter (GlyT) blocking are relevant for AAP’s action. Clozapine has a unique receptor profile on these molecular targets and therefore it is still considered the gold standard, especially for treatment-resistant schizophrenia. Recently, new concepts such as biased agonism and receptor dimerization have been introduced to explain the differences among different APs [19][20][21]. It has been found that some AAPs have biased signaling activities at D2 and 5-HT2A receptors, allowing them to preferentially block or activate specific receptor-mediated intracellular signaling pathways. For instance, clozapine has been shown to act as a biased agonist in vivo and in vitro at 5-HT2A receptor and to activate ERK and Akt kinases, and this can be another factor determining clozapine singularity [19][22].
3. Effects of AAPs on Hypothalamus and Peripheral Tissues
The AAPs-induced MetS is the consequence of a very complex and broad activity of these drugs on the CNS and peripheral organs, especially by interfering with the activity of GPCRs expressed in these tissues. In addition, if we consider how the CNS through hypothalamus deeply regulates the peripheral organs by altering the concentrations of neuromodulators and hormones in the blood system, the role of the CNS and each organ in the genesis of the AAPs-induced MetS becomes difficult to establish. Therefore, in order to understand the mechanism of MetS, especially in terms of hyperglycemia and dyslipidemia, it is desirable to analyze separately the contribution of the hypothalamus and each relevant organ targeted by AAPs.
3.1. AAPs and Hypothalamus
Hypothalamus is the main sensor of the nutrient concentrations in the blood, such as glucose, and it strongly intervenes by influencing glucose and lipid metabolism. Hypothalamus controls glucose and lipid homeostasis coordinating several organs such as the liver, pancreas, adipose tissue, and skeletal muscle, via the autonomic nervous system and the neuroendocrine system. Sympathetic stimulation increases noradrenaline and adrenaline levels, glucagon secretion, production of glucose and lipolysis, and it reduces insulin secretion leading to transitory blood glucose increase. Conversely, parasympathetic activation causes the opposite i.e., increases insulin secretion and inhibits production of glucose [23].
AAPs strongly interfere with hypothalamic centers activity by targeting monoaminergic GPCRs, thus altering descending and ascending autonomic system control. Monoaminergic neurons in the basal brain secrete dopamine, serotonin, noradrenaline and histamine influencing hypothalamic arcuate (ARC) and paraventricular nuclei (PVN) through the GPCRs expressed in these areas.
Among the GPCRs targeted by AAPs, antagonism at H1 receptor has been implicated in hyperphagia, weight gain and metabolic dysregulation [24]. Importantly, H1 seems relevant in controlling glucose and lipid homeostasis independent of weight gain (Figure 2). A significant association has been found between the genetic variants of H1 (rs346074-rs346070) and obesity [25].
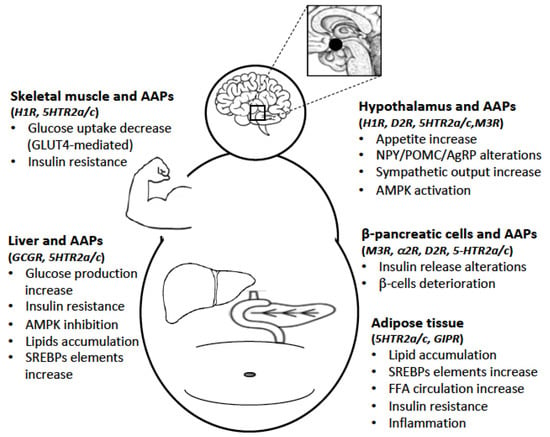
Figure 2. The AAPs-induced metabolic syndrome (MetS) is the consequence of a broad activity of these drugs on the central nervous system (CNS) and peripheral organs. In the CNS, the most important target is the hypothalamus, while in the periphery, the liver, pancreatic β-cells, adipose tissue and skeletal muscle are implicated in AAPs-induced MetS. By interfering with the activity of different GPCRs expressed in these regions, AAPs significantly alter glucose and lipid homeostasis.
A confirmation of the implication of H1 in the mechanism of AAPs comes from the observation that AAPs with high affinity for this receptor, such as clozapine and olanzapine, are the strongest for inducing weight gain and metabolic alterations. However, H1 antagonism is not the only mechanism involved in the hypothalamus because AAPs have a greater effect on MetS compared to selective antihistaminergic drugs [26]. In fact, dopaminergic and serotonergic antagonism of AAPs have a great impact on specific hypothalamic ARC neurons.
It is known that dopamine is relevant for food intake by regulating α-MSH and orexin expression, therefore D2 antagonism can cause an increase of hunger and thus weight gain. The ARC nucleus has a high concentration of D2 and D3 receptors [27], and their blockade can contribute to the deregulation of glucose and lipid metabolism. In addition, D2 antagonism induces hyperprolactinemia with consequences in food regulation and metabolism.
Regarding the role of serotonin in the hypothalamic centers, serotonergic neurons project to the pro-opiomelanocortin (POMC) centers of the ARC nucleus that express 5-HT2A/2C receptors, which reduce the appetite by increasing α-MSH secretion from POMC neurons that leads to melanocortin 4 receptors (MC4Rs) activation [28][29]. ARC nucleus is a relevant sensor for energetic homeostasis because it is sensitive to various neuro-humoral stimuli such as leptin, orexin, insulin, GLP-1, cholecystokinin and ghrelin [26].
Clozapine and olanzapine are potent 5-HT2A/2C antagonists and this is relevant for their effect on glucose and lipid metabolism, and weight gain [30]. In animal models, administration of selective 5-HT2A/2C antagonists resulted in significant increases in insulin resistance and insulin secretion in response to glucose stimulation. Conversely, 5-HT2C agonists have been proposed for the treatment of hyperglycemia and weight gain, and they have been experimented in clinical practice [31].
Alterations in hypothalamic orexigenic and anorexigenic neuropeptides expression and function, including neuropeptide Y (NPY), agouti-related protein (AgRP) and melanocortins have also been demonstrated during AAPs treatment, mainly due to their interference with monoaminergic systems.
In several experiments, it has been shown that AAPs, particularly clozapine and olanzapine, reduce the expression of the anorexigenic peptide POMC and conversely increase the expression of the orexigenic peptide AgRP and NPY [32].
As a consequence of these remarkable changes in the hypothalamus, AAPs increase the sympathetic outflow in the periphery by increasing glucagon secretion [33]. Enhanced levels of norepinephrine have been found in schizophrenic patients taking clozapine and risperidone, but not haloperidol [34]. As a confirmation of implication of the sympathetic system activation, the use of α2 or β-adrenergic receptor antagonist with clozapine was able to reverse hyperglycemia and reduce insulin secretion caused by clozapine [35].
Another important target whose function is altered during AAPs use is hypothalamic AMPK. AMPK, highly expressed in the ARC and ventromedial hypothalamus, is a key player in the regulation of energy homeostasis and metabolism, which is influenced by several neurotransmitters and neuropeptides through receptors that are targeted by AAPs [36]. When glucose levels decrease in the brain, AMPK is activated to replenish these levels that are essential for neuronal activity. Consequently, AMPK activates hepatic gluconeogenesis and glycogenolysis via stimulation of the sympathetic nervous system, which increases the secretion of hormones, such as glucagon, corticosterone and epinephrine [37]. AMPK activity is regulated by nutrients, anorexigenic (e.g., leptin) and orexigenic signals, and AAPs hinder these sensing mechanisms.
AAPs are known to increase hypothalamic AMPK activity mostly as antagonists at H1 receptors. In fact, AAPs with high affinity for H1, such as clozapine and olanzapine, are the strongest AMPK activators, whereas others, such as ziprasidone or lurasidone, have a minimal effect [38]. A study confirmed this hypothesis where clozapine was unable to activate hypothalamic AMPK phosphorylation in H1 KO mice, and thus its effect on food intake or weight gain disappeared in these mice [38]. On this line of evidence, the histaminergic stimulant betahistine partially reversed the changes in hypothalamic AMPK activation and NPY expression caused by olanzapine [36].
It should be noted that while AMPK activation in the hypothalamus increases glucose levels and alters lipid metabolism, the same mechanism has an opposite effect in the peripheral organs, such as the liver where it lowers glucose production and increases fatty acid oxidation.
3.2. AAPs and Liver
The liver is the principal location of glucose production, and it has a central role in the regulation of systemic glucose and lipid fluxes during feeding and fasting. Since the pancreatic veins drain directly into the portal venous system, every hormone secreted by the pancreas, such as insulin and glucagon, pass through the liver before entering into the systemic circulation. About 70% of hepatic glucose output takes place via glycogenolysis and the remaining 30% via gluconeogenesis [39].
The AAPs-induced increase of glucagon targets the liver that is the principal source of glucose production, and this excessive production is responsible for the development of T2D, even at early stages (Figure 2). In fact, clozapine and olanzapine may induce an increase in glucagon levels even when peripheral glucose levels are high [40]. Glucagon increases the expression of glucose 6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK) in the hepatocytes [41], which determine the rate of hepatic glucose production.
A confirmation of AAPs-induced glucagon increase for the hyperglycemic effect is based on early data where olanzapine-induced increase of blood glucose levels was found abolished in glucagon receptor KO mice [42]. Besides glucagon increase, the diabetogenic effect induced by AAPs such as clozapine and olanzapine is also mediated by hepatic insulin resistance as demonstrated in preclinical and clinical studies [43], which contributes to an abnormal increase of hepatic glucose output [40][44]. This effect was however not observed with risperidone [44]. The hepatic insulin resistance induced by clozapine can indeed occur rapidly following a single dose as demonstrated in healthy animals [35].
In relation to hepatic metabolism, AMPK is known to be a key player for glucose homeostasis because its activation lowers blood glucose levels, mostly by inhibiting gluconeogenesis. AAPs significantly decrease AMPK activity thereby altering glucose metabolism [45]. In addition, since activated AMPK also regulates the lipid metabolism by inhibiting lipogenesis and increasing fatty acid oxidation, it is reasonable to assume that AAPs increase hepatic lipogenesis through AMPK inhibition, which contributes to liver fat accumulation [46]. AMPK activation inhibits mTORc1 function, therefore AAPs indirectly stimulate mTOR signaling that increases the expression of the transcriptional activation of sterol-regulatory element-binding proteins SREBP-1c [46]. SREBPs play a central role in controlling a variety of lipid biosynthetic pathways, regulating genes for lipid and cholesterol biosynthesis. AAPs-induced hepatic overexpression of SREBP-1c is relevant for lipid accumulation and liver steatosis. These adverse effects have been demonstrated for clozapine, olanzapine, and risperidone, while aripiprazole or haloperidol did not induce the same effect [47].
SREBPs activity appears to be controlled by the downstream pathways of different receptors present in the liver, and among them, 5-HT2 and H1 have received particular attention, whose expression is increased in the liver of patients chronically exposed to AAPs [46][48].
In addition, it has been demonstrated that AAPs decrease the transcriptional activity of PPAR, another critical regulator of lipolysis and fatty acid oxidation in the liver but also in the adipose tissue [46].
Pharmacological treatments with antidiabetic drugs such as metformin and PPAR agonists reduce hepatic steatosis and glucose production through activation of AMPK and inhibition of SREBP elements, acting opposite to the mechanism of AAPs.
3.3. AAPs and Pancreatic β-Cells
AAPs treatment is associated with T2D, an undesired effect independent of weight gain. Considering that hyperglycemia and peripheral insulin resistance are common side effects induced by AAPs, a compensatory hyperinsulinemia should be expected regardless, and this makes difficult to establish the direct or indirect effects of AAPs on insulin secretion by the pancreatic β-cells. In fact, determining the effect of AAPs on β-cells activity is challenging because conflicting results have been reported. Few in vitro studies demonstrated that clozapine and olanzapine increase basal insulin secretion whereas haloperidol, ziprasidone or aripiprazole did not induce the same effect [49]. In one study, clozapine quickly increased basal insulin secretion at high concentrations; however, it did not alter glucose-stimulated insulin activity [50]. In vitro early studies proposed an inhibitory effect induced by some APs on GSIS [51].
These changes were later confirmed in clinical practice where 30–60% of patients using clozapine or olanzapine showed hyperinsulinemia; however, it is not clear whether this was a compensatory mechanism to insulin resistance or a direct consequence of β-cells stimulation by AAPs [50]. Besides, chronic hyperinsulinemia becomes an aggravating factor for β-cells deterioration and for early onset of T2D.
In relation to the mechanism of AAPs-induced hyperinsulinemia, D2, D3, 5-HT2A/2C and 5-HT1A receptors expressed in the β-cells might be partially responsible, considering that peripheral dopamine and serotonin generally inhibit insulin secretion in a subtle way (Figure 2). In fact, D2 antagonists, such as AAPs and TAPs, slightly increase insulin secretion, while dopamine agonists have an opposite effect [52]. Dopamine seems to be produced in the β-cells starting from its precursors present in the diet and/or from noradrenergic fibers, and in addition, it has a paracrine and autocrine inhibitory function on insulin secretion via D2 and D3 receptors [52].
Similarly, studies conducted on animal models demonstrated that serotonergic agonists and antagonists have an opposite effects on β-cells insulin secretion by acting on different serotonergic receptors such as 5-HT2A, 5-HT2C, and 5-HT1 [53].
Therefore, it is plausible that the combination of dopaminergic and serotonergic antagonism might contribute to AAPs-induced insulin hypersecretion, more than compounds that are selective at just one receptor subtype. Additionally, the antimuscarinic properties of some AAPs, such as clozapine and olanzapine, have an impact on the parasympathetic control of β-cells insulin secretion. On this aspect, the effect of AAPs is controversial because besides blocking muscarinic receptors, clozapine and olanzapine seem to increase the parasympathetic cholinergic output that probably leads to a rebound on the vagal system with compensatory stimulation of the β-cells [26][54]. In a study, olanzapine administrated for 9 days increased insulin and C peptide secretion that was reduced by atropine treatment, thus confirming the role of muscarinic receptor stimulation [55]. Clozapine and olanzapine-induced insulin dysregulation may also be partly due to blockade of hypothalamic M3 receptors [56].
Besides, the antagonism of clozapine at α2 receptors expressed on β-cells could reduce the inhibitory effect of the sympathetic system on insulin secretion, and thus have a role in hyperinsulinemia.
To demonstrate the difficulty in establishing definitive evidence on AAPs effect on β-cells insulin secretion, a recent in vitro study found that olanzapine actually reduces insulin secretion in the nanomolar concentration, whereas other works were conducted mostly at higher concentrations. The study proposed that blockade of D3, 5-HT2B, 5-HT2C and H1 receptors expressed in β-cells could be the mechanism responsible for AAPs-reduced insulin secretion [57].
In conclusion, the effect of AAPs on β-cells insulin secretion is still controversial and the hyperinsulinemia caused by AAPs as reported by several papers could be due to peripheral insulin resistance and/or β-cells overstimulation.
3.4. AAPs and Adipose Tissue
Data from animal and human studies have confirmed an increase of lipogenesis, visceral fat reserves, FFA circulation and differentiation of pre-adipocytes during AAPs treatment [58]. Studies on cultured cells have shown enhanced lipogenesis in rat adipocytes treated with olanzapine and clozapine [59]. In vivo and in vitro studies both have suggested that clozapine and olanzapine may increase lipogenesis through SREBP1c up-regulation [60], a transcription factor for several genes that are implicated in lipid metabolism (Figure 2).
Undesired changes in adiposity and insulin sensitivity were observed after 12 weeks of AAPs treatment in young patients, including greatest fat increase with olanzapine. In fact, several trials have shown that this phenomenon is more relevant in patients treated with olanzapine and clozapine compared to other AAPs [61][62].
In addition, AAPs differ from other drugs because they are capable of inducing hypertriglyceridemia. Generally, clozapine and olanzapine have the highest propensity to increase triglycerides levels, while quetiapine and risperidone are associated with moderate risk, whereas ziprasidone, lurasidone and aripiprazole seem to have a minimal risk [63].
Besides these effects, AAPs such as olanzapine and clozapine also increase plasma FFAs levels in patients and healthy persons. It is known that increased FFAs impair the ability of insulin to suppress hepatic glucose production and stimulate glucose uptake by skeletal muscle [64].
FFAs are generally sequestered from the plasma by hepatocytes, myocytes and adipocytes and then transformed into activated fatty acids, which are subsequently metabolized via oxidation or conserved via lipogenesis. When FFAs production exceeds the capacity of these two pathways to dispose them, then FFAs and their intermediates have a negative effect on insulin action at the cellular level. Some studies have illustrated that an excess of FFAs induces insulin receptor inactivation and degradation, including disruption of IRS1 [65][66].
Besides metabolic alterations, it has been demonstrated that AAPs are able to stimulate the differentiation of preadipocytes into adipocytes, thereby contributing to adipose tissue mass. In fact, olanzapine and clozapine were found to upregulate the expression of transcription factors essential for adipocyte differentiation [67]. Due to increased adiposity, a rise in leptin levels generally occurs, which in the long term can lead to leptin-resistance in the hypothalamic centers, thereby altering appetite regulation [68]. Recently, it was shown in 140 subjects that low adiponectin and high leptin levels in dysfunctional adipose tissues may contribute to increased oxidative stress and inflammation, which can be a marker for severity of MS [69].
All these above-mentioned alterations indicate the relevance of adipose tissue in the onset of MetS that causes insulin resistance, overstimulation of β-cells insulin secretion and inflammation, which in the long-term could lead to T2D and obesity.
3.5. AAPs and Skeletal Muscle
Based on the evidence that the H1 and 5-HT2A receptors play an important role in glycogen synthesis and glucose uptake mostly through GLUT-4 in muscle cells, it is reasonable to expect that glucose metabolism might be locally impacted by AAPs having strong antagonism at H1 and 5-HT2A, especially by reducing plasma glucose clearance that contributes to the onset of T2D [70] (Figure 2).
In an in vitro study, olanzapine inhibited IRS-1 and Akt phosphorylation induced by insulin and impaired insulin-signaling cascade, and this can be relevant for the peripheral insulin resistance [71]. Similar evidence was also found for clozapine in L6 muscle cells; however, ex vivo studies on rat-isolated skeletal muscle found contradictive results for AAPs [70].
Another factor that reduces skeletal muscle sensitivity to insulin is the increase of plasma FFA. As aforesaid for the adipose tissue, FFA can also interfere negatively with insulin receptor downstream signaling in the muscle cell and induce receptor degradation as well [66].
In addition, lipidomic studies on lipid contents (e.g., FFA, phosphatidylcholines and ceramides) in skeletal muscle biopsies from patients treated with AAPs have found several differences compared to the general population with potential consequences in insulin resistance; conversely, mood stabilizers did not cause the same shift [72].
References
- De Hert, M.A.; Van Winkel, R.; Van Eyck, D.; Hanssens, L.; Wampers, M.; Scheen, A.; Peuskens, J. Prevalence of the metabolic syndrome in patients with schizophrenia treated with antipsychotic medication. Schizophr. Res. 2006, 83, 87–93.
- Yevtushenko, O.O.; Cooper, S.J.; O’Neill, R.; Doherty, J.K.; Woodside, J.V.; Reynolds, G.P. Influence of 5-HT2C receptor and leptin gene polymorphisms, smoking and drug treatment on metabolic disturbances in patients with schizophrenia. Br. J. Psychiatry 2008, 192, 424–428.
- Laursen, T.M. Life expectancy among persons with schizophrenia or bipolar affective disorder. Schizophr. Res. 2011, 131, 101–104.
- Taipale, H.; Tanskanen, A.; Mehtälä, J.; Vattulainen, P.; Correll, C.U.; Tiihonen, J. 20-year follow-up study of physical morbidity and mortality in relationship to antipsychotic treatment in a nationwide cohort of 62,250 patients with schizophrenia (FIN20). World Psychiatry 2020, 19, 61–68.
- Tiihonen, J.; Wahlbeck, K.; Lönnqvist, J.; Klaukka, T.; Ioannidis, J.P.A.; Volavka, J.; Haukka, J. Effectiveness of antipsychotic treatments in a nationwide cohort of patients in community care after first hospitalisation due to schizophrenia and schizoaffective disorder: Observational follow-up study. Br. Med. J. 2006, 333, 224–227.
- Dickerson, F.B.; Brown, C.H.; Daumit, G.L.; Lijuan, F.; Goldberg, R.W.; Wohlheiter, K.; Dixon, L.B. Health status of individuals with serious mental illness. Schizophr. Bull. 2006, 32, 584–589.
- Aringhieri, S.; Carli, M.; Kolachalam, S.; Verdesca, V.; Cini, E.; Rossi, M.; McCormick, P.J.; Corsini, G.U.; Maggio, R.; Scarselli, M. Molecular targets of atypical antipsychotics: From mechanism of action to clinical differences. Pharmacol. Ther. 2018, 192, 20–41.
- Huhn, M.; Nikolakopoulou, A.; Schneider-Thoma, J.; Krause, M.; Samara, M.; Peter, N.; Arndt, T.; Bäckers, L.; Rothe, P.; Cipriani, A.; et al. Comparative efficacy and tolerability of 32 oral antipsychotics for the acute treatment of adults with multi-episode schizophrenia: A systematic review and network meta-analysis. Lancet 2019, 394, 939–951.
- Kowalchuk, C.; Castellani, L.N.; Chintoh, A.; Remington, G.; Giacca, A.; Hahn, M.K. Antipsychotics and glucose metabolism: How brain and body collide. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E1–E15.
- Ballon, J.S.; Pajvani, U.; Freyberg, Z.; Leibel, R.L.; Lieberman, J.A. Molecular pathophysiology of metabolic effects of antipsychotic medications. Trends Endocrinol. Metab. 2014, 25, 593–600.
- Gründer, G.; Hippius, H.; Carlsson, A. The “atypicality” of antipsychotics: A concept re-examined and re-defined. Nat. Rev. Drug Discov. 2009, 8, 197–202.
- Gillespie, A.L.; Samanaite, R.; Mill, J.; Egerton, A.; MacCabe, J.H. Is treatment-resistant schizophrenia categorically distinct from treatment-responsive schizophrenia? A systematic review. BMC Psychiatry 2017, 17, 1–14.
- Scarselli, M.; Armogida, M.; Chiacchio, S.; DeMontis, M.G.; Colzi, A.; Corsini, G.U.; Maggio, R. Reconstitution of functional dopamine D(2s) receptor by co-expression of amino- and carboxyl-terminal receptor fragments. Eur. J. Pharmacol. 2000, 397, 291–296.
- Rossi, M.; Fasciani, I.; Marampon, F.; Maggio, R.; Scarselli, M. The first negative allosteric modulator for dopamine D2 and D3 receptors, SB269652 may lead to a new generation of antipsychotic drugs. Mol. Pharmacol. 2017, 91, 586–594.
- Maggio, R.; Scarselli, M.; Capannolo, M.; Millan, M.J. Novel dimensions of D3 receptor function: Focus on heterodimerisation, transactivation and allosteric modulation. Eur. Neuropsychopharmacol. 2015, 25, 1470–1479.
- Fasciani, I.; Petragnano, F.; Aloisi, G.; Marampon, F.; Carli, M.; Scarselli, M.; Maggio, R.; Rossi, M. Allosteric modulators of g protein-coupled dopamine and serotonin receptors: A new class of atypical antipsychotics. Pharmaceuticals 2020, 13, 388.
- Meltzer, H.Y. Update on typical and atypical antipsychotic drugs. Annu. Rev. Med. 2013, 64, 393–406.
- Seeman, P. Targeting the dopamine D2 receptor in schizophrenia. Expert Opin. Ther. Targets 2006, 10, 515–531.
- Schmid, C.L.; Streicher, J.M.; Meltzer, H.Y.; Bohn, L.M. Clozapine acts as an agonist at serotonin 2A receptors to counter MK-801-induced behaviors through a βarrestin2-independent activation of akt. Neuropsychopharmacology 2014, 39, 1902–1913.
- Fasciani, I.; Pietrantoni, I.; Rossi, M.; Mannoury la Cour, C.; Aloisi, G.; Marampon, F.; Scarselli, M.; Millan, M.J.; Maggio, R. Distinctive binding properties of the negative allosteric modulator, [3H]SB269,652, at recombinant dopamine D3 receptors. Eur. J. Pharmacol. 2018, 819, 181–189.
- Carli, M.; Kolachalam, S.; Aringhieri, S.; Rossi, M.; Giovannini, L.; Maggio, R.; Scarselli, M. Dopamine D2 Receptors Dimers: How can we Pharmacologically Target Them? Curr. Neuropharmacol. 2017, 16, 222–230.
- Aringhieri, S.; Kolachalam, S.; Gerace, C.; Carli, M.; Verdesca, V.; Brunacci, M.G.; Rossi, C.; Ippolito, C.; Solini, A.; Corsini, G.U.; et al. Clozapine as the most efficacious antipsychotic for activating ERK 1/2 kinases: Role of 5-HT2A receptor agonism. Eur. Neuropsychopharmacol. 2017, 27, 383–398.
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of Insulin Synthesis and Secretion and Pancreatic Beta-Cell Dysfunction in Diabetes. Curr. Diabetes Rev. 2012, 9, 25–53.
- Tabarean, I.V. Histamine receptor signaling in energy homeostasis. Neuropharmacology 2016, 106, 13–19.
- Vehof, J.; Risselada, A.J.; Al Hadithy, A.F.Y.; Burger, H.; Snieder, H.; Wilffert, B.; Arends, J.; Wunderink, L.; Knegtering, H.; Wiersma, D.; et al. Association of genetic variants of the histamine H1 and muscarinic M3 receptors with BMI and HbA1c values in patients on antipsychotic medication. Psychopharmacology 2011, 216, 257–265.
- Reynolds, G.P.; Kirk, S.L. Metabolic side effects of antipsychotic drug treatment—Pharmacological mechanisms. Pharmacol. Ther. 2010, 125, 169–179.
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217.
- Xu, Y.; Jones, J.E.; Lauzon, D.A.; Anderson, J.G.; Balthasar, N.; Heisler, L.K.; Zinn, A.R.; Lowell, B.B.; Elmquist, J.K. A serotonin and melanocortin circuit mediates D-fenfluramine anorexia. J. Neurosci. 2010, 30, 14630–14634.
- Lam, D.D.; Garfield, A.S.; Marston, O.J.; Shaw, J.; Heisler, L.K. Brain serotonin system in the coordination of food intake and body weight. Pharmacol. Biochem. Behav. 2010, 97, 84–91.
- Zhou, L.; Sutton, G.M.; Rochford, J.J.; Semple, R.K.; Lam, D.D.; Oksanen, L.J.J.; Thornton-Jones, Z.D.; Clifton, P.G.; Yueh, C.Y.; Evans, M.L.; et al. Serotonin 2C Receptor Agonists Improve Type 2 Diabetes via Melanocortin-4 Receptor Signaling Pathways. Cell Metab. 2007, 6, 398–405.
- Bickerdike, M. 5-HT2C Receptor Agonists as Potential Drugs for the Treatment of Obesity. Curr. Top. Med. Chem. 2005, 3, 885–897.
- Fernø, J.; Varela, L.; Skrede, S.; Vázquez, M.J.; Nogueiras, R.; Diéguez, C.; Vidal-Puig, A.; Steen, V.M.; López, M. Olanzapine-induced hyperphagia and weight gain associate with orexigenic hypothalamic neuropeptide signaling without concomitant AMPK phosphorylation. PLoS ONE 2011, 6.
- Vatamaniuk, M.Z.; Horyn, O.V.; Vatamaniuk, O.K.; Doliba, N.M. Acetylcholine affects rat liver metabolism via type 3 muscarinic receptors in hepatocytes. Life Sci. 2003, 72, 1871–1882.
- Breier, A.; Wolkowitz, O.M.; Roy, A.; Potter, W.Z.; Pickar, D. Plasma norepinephrine in chronic schizophrenia. Am. J. Psychiatry 1990, 147, 1467–1470.
- Savoy, Y.E.; Ashton, M.A.; Miller, M.W.; Nedza, F.M.; Spracklin, D.K.; Hawthorn, M.H.; Rollema, H.; Matos, F.F.; Hajos-Korcsok, E. Differential effects of various typical and atypical antipsychotics on plasma glucose and insulin levels in the mouse: Evidence for the involvement of sympathetic regulation. Schizophr. Bull. 2010, 36, 410–418.
- Lian, J.; Huang, X.F.; Pai, N.; Deng, C. Betahistine ameliorates olanzapine-induced weight gain through modulation of histaminergic, NPY and AMPK pathways. Psychoneuroendocrinology 2014, 48, 77–86.
- Steinberg, G.R.; Kemp, B.E. AMPK in health and disease. Physiol. Rev. 2009, 89, 1025–1078.
- Kim, S.F.; Huang, A.S.; Snowman, A.M.; Teuscher, C.; Snyder, S.H. Antipsychotic drug-induced weight gain mediated by histamine H1 receptor-linked activation of hypothalamic AMP-kinase. Proc. Natl. Acad. Sci. USA 2007, 104, 3456–3459.
- Han, H.S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, 1–10.
- Smith, G.C.; Chaussade, C.; Vickers, M.; Jensen, J.; Shepherd, P.R. Atypical antipsychotic drugs induce derangements in glucose homeostasis by acutely increasing glucagon secretion and hepatic glucose output in the rat. Diabetologia 2008, 51, 2309–2317.
- Budick-Harmelin, N.; Anavi, S.; Madar, Z.; Tirosh, O. Fatty acids-stress attenuates gluconeogenesis induction and glucose production in primary hepatocytes. Lipids Health Dis. 2012, 11, 1–11.
- BONACCORSI, A.; GARATTINI, S.; JORI, A. Studies on the Hyperglycaemia Induced By Chlorpromazine in Rats. Br. J. Pharmacol. Chemother. 1964, 23, 93–100.
- Manu, P.; Correll, C.U.; Wampers, M.; van Winkel, R.; Yu, W.; Shiffeldrim, D.; Kane, J.M.; De Hert, M. Insulin secretion in patients receiving clozapine, olanzapine, quetiapine and risperidone. Schizophr. Res. 2013, 143, 358–362.
- Houseknecht, K.L.; Robertson, A.S.; Zavadoski, W.; Gibbs, E.M.; Johnson, D.E.; Rollema, H. Acute effects of atypical antipsychotics on whole-body insulin resistance in rats: Implications for adverse metabolic effects. Neuropsychopharmacology 2007, 32, 289–297.
- Oh, K.J.; Park, J.; Lee, S.Y.; Hwang, I.; Kim, J.B.; Park, T.S.; Lee, H.J.; Koo, S.H. Atypical antipsychotic drugs perturb AMPK-dependent regulation of hepatic lipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2011, 300, 624–632.
- Xu, H.; Zhuang, X. Atypical antipsychotics-induced metabolic syndrome and nonalcoholic fatty liver disease: A critical review. Neuropsychiatr. Dis. Treat. 2019, 15, 2087–2099.
- Cai, H.L.; Tan, Q.Y.; Jiang, P.; Dang, R.L.; Xue, Y.; Tang, M.M.; Xu, P.; Deng, Y.; Li, H.D.; Yao, J.K. A potential mechanism underlying atypical antipsychotics-induced lipid disturbances. Transl. Psychiatry 2015, 5.
- Siafis, S.; Tzachanis, D.; Samara, M.; Papazisis, G. Antipsychotic Drugs: From Receptor-binding Profiles to Metabolic Side Effects. Curr. Neuropharmacol. 2017, 16, 1210–1223.
- Melkersson, K.; Khan, A.; Hilding, A.; Hulting, A.L. Different effects of antipsychotic drugs on insulin release in vitro. Eur. Neuropsychopharmacol. 2001, 11, 327–332.
- Melkersson, K. Clozapine and olanzapine, but not conventional antipsychotics, increase insulin release in vitro. Eur. Neuropsychopharmacol. 2004, 14, 115–119.
- Sussman, K.E.; Pollard, H.B.; Leitner, J.W.; Nesher, R.; Adler, J.; Cerasi, E. Differential control of insulin secretion and somatostatin-receptor recruitment in isolated pancreatic islets. Biochem. J. 1983, 214, 225–230.
- Rubi, B.; Ljubicic, S.; Pournourmohammadi, S.; Carobbio, S.; Armanet, M.; Bartley, C.; Maechler, P. Dopamine D2-like receptors are expressed in pancreatic beta cells and mediate inhibition of insulin secretion. J. Biol. Chem. 2005, 280, 36824–36832.
- Zhang, Q.; Zhu, Y.; Zhou, W.; Gao, L.; Yuan, L.; Han, X. Serotonin Receptor 2C and Insulin Secretion. PLoS ONE 2013, 8.
- Teff, K.L.; Rickels, M.R.; Grudziak, J.; Fuller, C.; Nguyen, H.L.; Rickels, K. Antipsychotic-induced insulin resistance and postprandial hormonal dysregulation independent of weight gain or psychiatric disease. Diabetes 2013, 62, 3232–3240.
- Rickels, M.R.; Perez, E.M.; Peleckis, A.J.; Alshehabi, E.; Nguyen, H.L.; Stefanovski, D.; Rickels, K.; Teff, K.L. Contribution of parasympathetic muscarinic augmentation of insulin secretion to olanzapine-induced hyperinsulinemia. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E250–E257.
- Weston-Green, K.; Huang, X.F.; Deng, C. Second generation antipsychotic-induced type 2 diabetes: A role for the muscarinic M3 receptor. CNS Drugs 2013, 27, 1069–1080.
- Nagata, M.; Yokooji, T.; Nakai, T.; Miura, Y.; Tomita, T.; Taogoshi, T.; Sugimoto, Y.; Matsuo, H. Blockade of multiple monoamines receptors reduce insulin secretion from pancreatic β-cells. Sci. Rep. 2019, 9, 1–10.
- Gonçalves, P.; Araújo, J.R.; Martel, F. Antipsychotics-induced metabolic alterations: Focus on adipose tissue and molecular mechanisms. Eur. Neuropsychopharmacol. 2015, 25, 1–16.
- Vestri, H.S.; Maianu, L.; Moellering, D.R.; Garvey, W.T. Atypical antipsychotic drugs directly impair insulin action in adipocytes: Effects on glucose transport, lipogenesis, and antilipolysis. Neuropsychopharmacology 2007, 32, 765–772.
- Rojo, L.E.; Gaspar, P.A.; Silva, H.; Risco, L.; Arena, P.; Cubillos-Robles, K.; Jara, B. Metabolic syndrome and obesity among users of second generation antipsychotics: A global challenge for modern psychopharmacology. Pharmacol. Res. 2015, 101, 74–85.
- Albaugh, V.L.; Singareddy, R.; Mauger, D.; Lynch, C.J. A double blind, placebo-controlled, randomized crossover study of the acute metabolic effects of olanzapine in healthy volunteers. PLoS ONE 2011, 6.
- Nicol, G.E.; Yingling, M.D.; Flavin, K.S.; Schweiger, J.A.; Patterson, B.W.; Schechtman, K.B.; Newcomer, J.W. Metabolic effects of antipsychotics on adiposity and insulin sensitivity in youths a randomized clinical trial. JAMA Psychiatry 2018, 75, 788–796.
- Pillinger, T.; McCutcheon, R.A.; Vano, L.; Mizuno, Y.; Arumuham, A.; Hindley, G.; Beck, K.; Natesan, S.; Efthimiou, O.; Cipriani, A.; et al. Comparative effects of 18 antipsychotics on metabolic function in patients with schizophrenia, predictors of metabolic dysregulation, and association with psychopathology: A systematic review and network meta-analysis. The Lancet Psychiatry 2020, 7, 64–77.
- Jassim, G.; Skrede, S.; Vázquez, M.J.; Wergedal, H.; Vik-Mo, A.O.; Lunder, N.; Diéguez, C.; Vidal-Puig, A.; Berge, R.K.; López, M.; et al. Acute effects of orexigenic antipsychotic drugs on lipid and carbohydrate metabolism in rat. Psychopharmacology 2012, 219, 783–794.
- Gao, Z.; Zhang, X.; Zuberi, A.; Hwang, D.; Quon, M.J.; Lefevre, M.; Ye, J. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol. Endocrinol. 2004, 18, 2024–2034.
- Schenk, S.; Saberi, M.; Olefsky, J.M. Insulin sensitivity: Modulation by nutrients and inflammation. J. Clin. Investig. 2008, 118, 2992–3002.
- Hemmrich, K.; Gummersbach, C.; Pallua, N.; Luckhaus, C.; Fehsel, K. Clozapine enhances differentiation of adipocyte progenitor cells. Mol. Psychiatry 2006, 11, 980–981.
- Barateiro, A.; Mahú, I.; Domingos, A.I. Leptin resistance and the neuro-adipose connection. Front. Endocrinol. 2017, 8, 8–11.
- Frühbeck, G.; Catalán, V.; Rodríguez, A.; Ramírez, B.; Becerril, S.; Salvador, J.; Portincasa, P.; Colina, I.; Gómez-Ambrosi, J. Involvement of the leptin-adiponectin axis in inflammation and oxidative stress in the metabolic syndrome. Sci. Rep. 2017, 7, 1–8.
- Grajales, D.; Ferreira, V.; Valverde, Á.M. Second-Generation Antipsychotics and Dysregulation of Glucose Metabolism: Beyond Weight Gain. Cells 2019, 8, 1336.
- Ren, L.; Zhou, X.; Huang, X.; Wang, C.; Li, Y. The IRS/PI3K/Akt signaling pathway mediates olanzapine-induced hepatic insulin resistance in male rats. Life Sci. 2019, 217, 229–236.
- Burghardt, K.J.; Ward, K.M.; Sanders, E.J.; Howlett, B.H.; Seyoum, B.; Yi, Z. Atypical antipsychotics and the human skeletal muscle lipidome. Metabolites 2018, 8, 64.

