 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Antonio Giovanni Solimando | + 1742 word(s) | 1742 | 2020-05-12 10:11:38 | | | |
| 2 | Catherine Yang | Meta information modification | 1742 | 2020-05-14 05:51:01 | | | | |
| 3 | Catherine Yang | -4 word(s) | 1738 | 2020-10-28 08:01:23 | | |
Video Upload Options
Immune checkpoint inhibitor (ICI)-related inflammatory diseases, including polymyositis (PM) and dermatomyositis (DM), in patients suffering from neoplastic disorders represent a medical challenge. The treatment of these conditions has taken on new urgency due to the successful and broad development of cancer-directed immunological-based therapeutic strategies. While primary and secondary PM/DM phenotypes have been pathophysiologically characterized, a rational, stepwise approach to the treatment of patients with ICI-related disease is lacking. In the absence of high-quality evidence to guide clinical judgment, the available data must be critically assessed. In this literature review, we examine partially neglected immunological and clinical findings to obtain insights into the biological profiles of ICI-related PM/DM and potential treatment options. We show that differential diagnosis is essential to stratifying patients according to prognosis and therapeutic impact. Finally, we provide a comprehensive assessment of druggable targets and suggest a stepwise patient-oriented approach for the treatment of ICI-related PM/DM.
1. Introduction
Immune surveillance has emerged as a pivotal issue in the invasiveness of both visceral [1] and skeletal [2][3] malignancies, as it fuels a vicious cycle between the neoplastic cells and the immune microenvironment. Immune checkpoint inhibitor (ICI)-based approaches, aimed at interrupting corrupted immune bystander cells and reactivating an effective anti-cancer response, represent one of the most significant therapeutic innovations in the oncologic landscape to date [4] with an ability to target solid [5][6] and hematological [7][8] malignancies. However, among the side effects of ICI therapy is ICI-related polymyositis (PM), an inflammatory process affecting the skeletal muscles. While this condition is rare, it can be severe and potentially deadly, as it may cause rhabdomyolysis in striated muscle, including the myocardium. PM can occur as a reactivation of a previous paraneoplastic polymyositis or dermatomyositis (DM) or as a new entity [9][10]. Clinically, PM/DM manifests as worsening muscle weakness and myalgias. Compared to non-ICI-related forms of inflammatory myositis, oculomotor and axial muscle involvement, including diplopia and muscular weakness as suggestive symptoms, have been reported [11][12][13][14]. An involvement of the bulbar musculature can cause dysarthria, dysphonia or dysphagia. Physical examination will reveal skin signs suggestive of DM, while a careful history can rule out alternative causes, such as chronic steroid myopathy. Blood chemistry tests support the diagnosis by detecting elevated serum levels of muscle damage markers (creatine phosphokinase, lactate dehydrogenase, transaminases, aldolases) and in some patients myositis-specific or myositis-related antibodies. ICI-therapy-related variants are characterized by the frequent involvement of other targets of the peripheral nervous system and of the myocardium (myasthenia gravis [MG], polyradiculoneuritis, myocarditis), which can be detected using specific tests: increased troponin levels suggest cardiac involvement; an electromyoneurography (EMG/ENG) study can confirm the presence of myogenic damage or the presence of neuropathic damage or neuromuscular plaque disease. Further information might be obtained with magnetic resonance imaging (MRI) of the muscle and/or muscle biopsy; the latter can identify secondary manifestations, such as giant cell arteritis, systemic lupus, and sarcoidosis.
In a meta-analysis of adverse events related to ICI, the incidence of grade 3–5 adverse events involving the central nervous system (encephalitis, encephalopathy, aseptic meningitis or myelitis) was 0.46% (22 of 4775 ICI-treated patients in 12 studies). In the same analysis, the incidence of peripheral neuropathy of any degree was 5% (220 of 4390 patients exposed to ICI in 17 studies), significantly lower than that occurring in association with conventional chemotherapy. Among 3128 patients from eight studies, the meta-analysis found four cases of grade 3–5 MG (0.13%) and three of grade 3–5 myositis (0.10%) [15]. However, the clinical scenario is often multifaced, being associated with several other uncommon features. Indeed, the presence of bulbar symptoms, dysphagia, or ocular motor symptoms can suggest the diagnosis of MG that it is not confirmed by antibody levels and/or nerve stimulation tests. A small portion have also cardiac myositis, which turns out to be among the most fatal of all the irAEs. Any of these three compartments of myasthenia-skeletal muscle myositis or carditis can occur individually or all together, variably impacting the patient outcome.
2. Biological Background: Bridging the Gaps between Immune Checkpoint Inhibition and Physiopathology ICI-Related Disease
Among the functions of the human immune system is tumor surveillance. In this stepwise response, antigen-presenting cells (APCs) control the tumoral antigen load by priming and activating T cells, which in turn recognize and then destroy the malignancy, thus releasing an even higher tumoral antigen load, which again elicits an immune response. However, when a single step in this process is impaired, the tumor can grow exponentially. Immune-directed therapy attempts to restore the homeostatic equilibrium [16]. One of the pharmacodynamically most successful ICI-related approaches to enhancing the anti-tumor immune response consists of interfering with the negative costimulation of T cells, by inhibiting PD-1, PD-L1, and CTLA-4 [17]. PD-1 (CD279) is a type I transmembrane receptor expressed on the surfaces of T cells, B cells, monocytes, natural killer cells, and dendritic cells. It has two physiological ligands: PD-L1 (B7-H1, CD274) and PD-L2 (B7-DC, CD273). These Ig-like transmembrane receptors are also expressed on the cell surface [17]. CTLA-4 is a CD28 homolog with a high affinity for B7-1/2. The binding of CTLA-4 to B7-1/2 acts as a co-inhibitory signal that hinders early T cell activation [18]. A study in CTLA-4-deficient animals demonstrated the pivotal role of CTLA-4 in halting T-cell-mediated immune anti-tumor activity [19]. Inborn immunity errors of immune checkpoints similarly predispose patients to autoimmune manifestations. For example, in CHAI/LATAIE, a type of CTLA-4 insufficiency, the clinical features mirror those of ipilimumab-related responses [19]. ICI modulators interrupt T cells and APCs, but also cancer cells. The immune checkpoints involved in these processes are also relevant in the pathogenesis and treatment of rheumatic diseases. The difference is that while in cancer therapy the negative stimulation is halted by ICI administration, in the treatment of autoimmunity, negative costimulation is promoted [9].
However, in the anti-tumoral activity of ICIs, a normal inflammatory reaction is elicited that can lead to immune-related adverse events (irAEs). The biological mechanisms by which those adverse events take place are still poorly understood [13] but several pathophysiological pathways have been proposed (Figure 1).
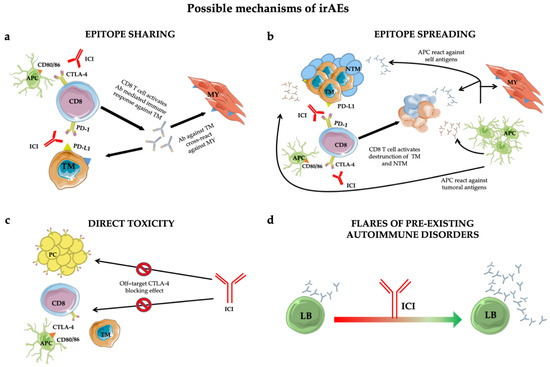
Figure 1. Mechanisms of immune-related adverse events (irAEs): epitope-sharing (a); -spreading (b); direct toxicity (c) and flares of pre-existing autoimmune disorders (d). See text for details. Abbreviations: ICI = immune checkpoint inhibitor; TM = tumoral cell; MY = myocytes; Ab = antibody; NTM = non tumoral cell; APC = antigen presenting cell; PC = pituitary cell; LB = B lymphocyte.
Treg cells are responsible for self-tolerance and constitutively express CTLA-4 on their surfaces [20]. By triggering antibody-dependent cellular cytotoxicity (ADCC)-mediated Treg depletion, CTLA-4 antagonists increase the T effector (Teff) cell proliferation and thus an immune response to the tumor [21]. PD-L1 antibodies break the link between PD-L1 and Treg, thus also reducing Treg generation. Depending on the corrupted equilibrium between Treg and Teff cells, a loss of peripheral tolerance induced by ICIs can drive not only the desired anti-tumor immune response but also autoimmune adverse events [21].
Additionally, the epitope-sharing (Figure 1a) between tumor and healthy tissue was shown to be the cause of a cross-reaction inducing Teff activation against self-tissues, evidenced in a patient who developed a lymphocyte infiltrate in heart and skeletal muscle upon ICI treatment [22].
An alternative mechanism leading to irAEs is represented by the epitope spreading (Figure 1b) [23], in which Teff-cell-mediated tumor cell death in the cancer-microenvironment causes the release of a huge amount of tumor antigens and self antigens. The resulting autoimmune reaction induces an uncontrolled propagation of this response [18].
Furthermore, autoptic evidence suggests that direct toxicity (Figure 1c) [24] can also trigger irAEs. This was shown in a subset of adenohypophyseal endocrine cells whose CTLA-4 expression mirrorred that of T cells. Consequently, therapeutic administration of an anti-CTLA-4 monoclonal antibody with antitumor effects could elicit type II and type IV hypersensitivity and hypophysitis.
Finally, preclinical predisposition to autoimmunity constitutes an exacerbation-initiating factor per se (Figure 1d) [25]. Patients with rheumatoid factor and auto-antibody positivity without clinical manifestations before treatment are more likely to develop irAEs. However, insights into the true impact of autoimmune substrates are lacking due to the exclusion from ICI trials of patients with autoimmune diseases. However, retrospective case series found no significant difference in the incidence of flares in patients with and without pre-existing autoimmune disorders [26][27][28][29][30].
3. Characteristic Autoantibody Patterns
The antibodies characteristic of inflammatory myopathies [31] are conventionally divided into those that are myositis-associated (MAAs) vs. myositis-specific (MSAs) [32]. Antibodies in the first group are a common feature of overlap syndromes involving other systemic autoimmune diseases [33], particularly scleroderma and systemic sclerosis. The best characterized antibodies are those that recognize the 52-kDa Ro/SSA antigen, a ribonucleoprotein associated with a complex that binds small RNA molecules; the DNAPK antigen (Ku/DNA-dependent protein kinase), a kinase necessary for DNA repair; and PM-Scl, a complex of 11 proteins with nucleolar localization that comprises the autoantigen of PM/scleroderma. The second group includes antibodies directed against histidyl transfer-RNA-synthetase (Jo-1) [34], the most frequently occurring antibodies. The latter are among a group of eight autoantibodies directed against aminoacyl-transfer-RNA-synthetase, which catalyses the binding of specific amino acids to transfer RNA. The presence of these autoantibodies identifies a subset of patients suffering from “anti-synthetase syndrome”. Autoantibodies that bind the SRP (signal recognition particle) are also members of the second group. SRP is an RNA-protein cytoplasmic complex that recognizes secreted and membrane-associated proteins in addition to regulating the translocation of proteins through the endoplasmic reticulum. Anti-SRP autoantibodies are associated with particularly severe myositis, extensive necrosis, and an unfavorable prognosis. Anti-Mi-2 antibodies recognize a helicase that is part of the NuRD (nucleosome remodeling deacetylase) complex, which plays a key role in gene transcription. While anti-Mi-2 antibodies are very specific for DM and generally associated with a favorable prognosis, they also increase the risk of cancer [35].
DM is associated with several antibodies targeting melanoma differentiation antigen 5 (MDA5) [36] and transcriptional intermediary factor 1 (TIF1) [37]. Patients positive for anti-MDA5 antibodies often have high-grade palmar rash, digital ulcers, rapidly progressive interstitial lung involvement, and amyopathic DM [38] whereas those with anti-TIF1 and anti-NXP2 antibodies have an increased risk of malignancies [39].
In most of cases with ICI-related myositis, MSAs or MAAs were undetected. Nevertheless, in a subgroup of ICI-treated patients, enrolled in clinical trials, banked serum demonstrated pretreatment auto-antibody positivity. Since the presence of asymptomatic autoantibody, upon ICI treatment, can be followed by an explosive disease, an asymptomatic phenotype might constitute the early irAEs phase in subjects genetically predisposed to full-blown disease [40][41]. In the frame of this thinking, ICI candidate baseline screening might contribute to detect autoimmune predisposition. To this end, real life studies might be worthy, since most patients with autoimmune disease were censored from clinical trials.
References
- Olga S. Blomberg; Lorenzo Spagnuolo; Karin E. De Visser; Immune regulation of metastasis: mechanistic insights and therapeutic opportunities. Disease Models & Mechanisms 2018, 11, dmm036236, 10.1242/dmm.036236.
- Gnoni Antonio; Brunetti Oronzo; Longo Vito; Calabrese Angela; Argentiero Antonel-La; Calbi Roberto; Solimando Antonio Giovanni; Licchetta Antonella; Immune system and bone microenvironment: rationale for targeted cancer therapies. Oncotarget 2020, 11, 480-487, 10.18632/oncotarget.27439.
- Antonella Argentiero; Antonio Giovanni Solimando; Oronzo Brunetti; Angela Calabrese; Francesco Pantano; Michele Iuliani; Daniele Santini; Nicola Silvestris; Angelo Vacca; Skeletal Metastases of Unknown Primary: Biological Landscape and Clinical Overview.. Cancers 2019, 11, 1270, 10.3390/cancers11091270.
- Saїd C Azoury; David M Straughan; Vivek Shukla; Immune Checkpoint Inhibitors for Cancer Therapy: Clinical Efficacy and Safety.. Current Cancer Drug Targets 2015, 15, 452-462, 10.2174/156800961506150805145120.
- Vito Longo; Oronzo Brunetti; Antonio Gnoni; Antonella Licchetta; Sabina Delcuratolo; R. Memeo; Antonio Giovanni Solimando; Antonella Argentiero; Emerging role of Immune Checkpoint Inhibitors in Hepatocellular Carcinoma. Medicina 2019, 55, 698, 10.3390/medicina55100698.
- Mark N. Stein; Jyoti Malhotra; Rohinton S. Tarapore; Usha Malhotra; Ann W. Silk; Nancy Chan; Lorna Rodriguez; Joseph Aisner; Robert D. Aiken; Tina Mayer; Bruce G. Haffty; Jenna H. Newman; Salvatore M. Aspromonte; Praveen K. Bommareddy; Ricardo Estupinian; Charles B. Chesson; Evita T. Sadimin; Shengguo Li; Daniel J. Medina; Tracie Saunders; Melissa Frankel; Aparna Kareddula; Sherrie Damare; Elayne Wesolowsky; Christian Gabel; Wafik S. El-Deiry; Varun V. Prabhu; Joshua E. Allen; Martin Stogniew; Wolfgang Oster; Joseph R. Bertino; Steven K. Libutti; Janice M. Mehnert; Andrew Zloza; Safety and enhanced immunostimulatory activity of the DRD2 antagonist ONC201 in advanced solid tumor patients with weekly oral administration. Journal for ImmunoTherapy of Cancer 2019, 7, 136, 10.1186/s40425-019-0599-8.
- Philippe Armand; Immune checkpoint blockade in hematologic malignancies. Blood 2015, 125, 3393-3400, 10.1182/blood-2015-02-567453.
- Patrizia Leone; Giuseppe Di Lernia; Antonio Giovanni Solimando; Sebastiano Cicco; Ilaria Saltarella; Aurelia Lamanuzzi; Roberto Ria; Maria Antonia Frassanito; Maurilio Ponzoni; Paolo Ditonno; Franco Dammacco; Vito Racanelli; Angelo Vacca; Bone marrow endothelial cells sustain a tumor-specific CD8+ T cell subset with suppressive function in myeloma patients. OncoImmunology 2018, 8, e1486949-12, 10.1080/2162402x.2018.1486949.
- Laura C. Cappelli; A. K. Gutierrez; Clifton O. Bingham; Ami A. Shah; Rheumatic and Musculoskeletal Immune-Related Adverse Events Due to Immune Checkpoint Inhibitors: A Systematic Review of the Literature.. Arthritis & Rheumatism 2017, 69, 1751-1763, 10.1002/acr.23177.
- Mehmet Asim Bilen; Sumit K. Subudhi; Jianjun Gao; Nizar M. Tannir; Shi-Ming Tu; Barbara Seliger; Acute rhabdomyolysis with severe polymyositis following ipilimumab-nivolumab treatment in a cancer patient with elevated anti-striated muscle antibody. Journal for ImmunoTherapy of Cancer 2016, 4, 36, 10.1186/s40425-016-0139-8.
- Mehdi Touat; Thierry Maisonobe; Samuel Knauss; Omar Ben Hadj Salem; Baptiste Hervier; Karine Auré; Tali-Anne Szwebel; Nora Kramkimel; Claire Lethrosne; Jean-Frédéric Bruch; Pauline Laly; Jacques Cadranel; Nicolas Weiss; Anthony Béhin; Y. Allenbachyves; Olivier Benveniste; Timothée Lenglet; Dimitri Psimaras; Werner Stenzel; Sarah Léonard-Louis; Immune checkpoint inhibitor-related myositis and myocarditis in patients with cancer. Neurology 2018, 91, e985-e994, 10.1212/wnl.0000000000006124.
- Shiro Matsubara; Morinobu Seki; Shigeaki Suzuki; Takashi Komori; Mikio Takamori; Tertiary lymphoid organs in the inflammatory myopathy associated with PD-1 inhibitors. Journal for ImmunoTherapy of Cancer 2019, 7, 256-7, 10.1186/s40425-019-0736-4.
- Mehdi Touat; Daniel Talmasov; Damien Ricard; Dimitri Psimaras; Neurological toxicities associated with immune-checkpoint inhibitors. Current Opinion in Neurology 2017, 30, 659-668, 10.1097/wco.0000000000000503.
- Justin C. Kao; Bing Liao; Svetomir N. Markovic; C J Klein; Elie Naddaf; Nathan P. Staff; Teerin Liewluck; Julie E. Hammack; Paola Sandroni; Heidi Finnes; Michelle L. Mauermann; Neurological Complications Associated With Anti-Programmed Death 1 (PD-1) Antibodies.. JAMA Neurology 2017, 74, 1216-1222, 10.1001/jamaneurol.2017.1912.
- Xu, M.; Nie, Y.; Yang, Y.; Lu, Y.-T.; Su, Q.; Risk of Neurological Toxicities Following the Use of Different Immune Checkpoint Inhibitor Regimens in Solid Tumors: A Systematic Review and Meta-analysis. Neurologist 2019, 24, 75–83.
- Daniel Chen; Ira Mellman; Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1-10, 10.1016/j.immuni.2013.07.012.
- Krzysztof M. Zak; Przemysław Grudnik; Katarzyna Magiera-Mularz; Alexander Dömling; Grzegorz Dubin; Tad A. Holak; Structural Biology of the Immune Checkpoint Receptor PD-1 and Its Ligands PD-L1/PD-L2. Structure 2017, 25, 1163-1174, 10.1016/j.str.2017.06.011.
- Ribas, A.; Wolchok, J.D.; Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355.
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P.; CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594.
- Iwona Kwiecień; Anna Stelmaszczyk-Emmel; Malgorzata Polubiec-Kownacka; Dariusz Dziedzic; Joanna Domagała-Kulawik; Elevated regulatory T cells, surface and intracellular CTLA-4 expression and interleukin-17 in the lung cancer microenvironment in humans.. Cancer Immunology Immunotherapy 2016, 66, 161-170, 10.1007/s00262-016-1930-6.
- Prabhakaran Kumar; Palash Bhattacharya; Bellur S. Prabhakar; A comprehensive review on the role of co-signaling receptors and Treg homeostasis in autoimmunity and tumor immunity. Journal of Autoimmunity 2018, 95, 77-99, 10.1016/j.jaut.2018.08.007.
- Maurie Markman; Johnson Db; Balko Jm; Compton Ml; Chalkias S; Gorham J; Xu Y; Hicks M; Puzanov I; Alexander; et al.Bloomer TlBecker JrSlosky DaPhillips EjPilkinton MaCraig-Owens LKola NPlautz GReshef DsDeutsch JsDeering RpOlenchock BaLichtman AhRoden DmSeidman CeKoralnik IjSeidman JgHoffman RdTaube JmDiaz LaAnders RaSosman JaMoslehi Jj Faculty Opinions recommendation of Fulminant Myocarditis with Combination Immune Checkpoint Blockade.. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2018, 375, 1749–1755, 10.3410/f.726914491.793541846.
- Carl H. June; Jeremy T Warshauer; Jeffrey A Bluestone; Is autoimmunity the Achilles' heel of cancer immunotherapy?. Nature Medicine 2017, 23, 540-547, 10.1038/nm.4321.
- Patrizio Caturegli; Giulia Di Dalmazi; Martina Lombardi; Federica Grosso; H. Benjamin Larman; Tatianna Larman; Giacomo Taverna; Mirco Cosottini; Isabella Lupi; Hypophysitis Secondary to Cytotoxic T-Lymphocyte-Associated Protein 4 Blockade: Insights into Pathogenesis from an Autopsy Series.. The American Journal of Pathology 2016, 186, 3225-3235, 10.1016/j.ajpath.2016.08.020.
- Yukihiro Toi; Shunichi Sugawara; Jun Sugisaka; Hirotaka Ono; Yosuke Kawashima; Tomoiki Aiba; Sachiko Kawana; Ryohei Saito; Mari Aso; Kyoji Tsurumi; et al.Kana SuzukiHisashi ShimizuYutaka DomekiKeisuke TerayamaAtsushi NakamuraShinsuke YamandaYuichiro KimuraYoshihiro Honda Profiling Preexisting Antibodies in Patients Treated With Anti-PD-1 Therapy for Advanced Non-Small Cell Lung Cancer.. JAMA Oncology 2018, 5, 376, 10.1001/jamaoncol.2018.5860.
- Stephen Ansell; Johnson Db; Sullivan Rj; Ott Pa; Carlino Ms; Khushalani Ni; Ye F; Guminski A; Puzanov I; Lawrence Dp; Buchbinder Ei; Mudigonda T; Spencer K; Bender C; Lee J; Kaufman Hl; Menzies Am; Hassel Jc; Mehnert Jm; Sosman Ja; Long Gv; Clark Ji; Faculty Opinions recommendation of Ipilimumab therapy in patients with advanced melanoma and preexisting autoimmune disorders.. Faculty Opinions – Post-Publication Peer Review of the Biomedical Literature 2016, 2, , 10.3410/f.725984091.793517172.
- Alexander M Menzies; D. B. Johnson; S. Ramanujam; V. G. Atkinson; A. N. M. Wong; John Park; J. L. McQuade; A. N. Shoushtari; K. K. Tsai; Z. Eroglu; O. Klein; J.C. Hassel; J.A. Sosman; A. Guminski; R. J. Sullivan; A. Ribas; M. S. Carlino; M. A. Davies; Shahneen Sandhu; Georgina V. Long; Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Annals of Oncology 2017, 28, 368-376, 10.1093/annonc/mdw443.
- Ralf Gutzmer; Anika Koop; Friedegund Meier; Jessica C. Hassel; Patrick Terheyden; Lisa Zimmer; Lucie Heinzerling; Selma Ugurel; Claudia Pföhler; Anja Gesierich; Elisabeth Livingstone; Imke Satzger; Katharina C. Kähler; Programmed cell death protein-1 (PD-1) inhibitor therapy in patients with advanced melanoma and preexisting autoimmunity or ipilimumab-triggered autoimmunity. European Journal of Cancer 2017, 75, 24-32, 10.1016/j.ejca.2016.12.038.
- Giulia C. Leonardi; Justin F. Gainor; Mehmet Altan; Sasha Kravets; Suzanne E. Dahlberg; Lydia Gedmintas; Roxana Azimi; Hira Rizvi; Jonathan W. Riess; Matthew D. Hellmann; Mark M. Awad; Safety of Programmed Death–1 Pathway Inhibitors Among Patients With Non–Small-Cell Lung Cancer and Preexisting Autoimmune Disorders. Journal of Clinical Oncology 2018, 36, 1905-1912, 10.1200/jco.2017.77.0305.
- Alice Tison; Gilles Quéré; Laurent Misery; Elisa Funck‐Brentano; François‐Xavier Danlos; Emilie Routier; Caroline Robert; Yohann Loriot; Olivier Lambotte; Bertille Bonniaud; Camille Scalbert; Sarah Maanaoui; Thierry Lesimple; Stéphanie Martinez; Marie Marcq; Christos Chouaid; Catherine Dubos; Florence Brunet‐Possenti; Chloé Stavris; Laurent Chiche; Nathalie Beneton; Sandrine Mansard; Florian Guisier; Hélène Doubre; François Skowron; François Aubin; Ouidad Zehou; Christophe Roge; Mickaël Lambert; Anne Pham‐Ledard; Marie Beylot‐Barry; Rémi Veillon; Nora Kramkimel; Damien Giacchero; Julie De Quatrebarbes; Catherine Michel; Jean‐Bernard Auliac; Gilles Gonzales; Chantal Decroisette; Gwenaelle Le Garff; Ioana Carpiuc; Hervé Vallerand; Emmanuel Nowak; Divi Cornec; Marie Kostine; Groupe de Cancérologie Cutanée, Groupe Français de Pneumo-Cancérologie, and Club Rhumatismes et Inflammations; Safety and Efficacy of Immune Checkpoint Inhibitors in Patients With Cancer and Preexisting Autoimmune Disease: A Nationwide, Multicenter Cohort Study. Arthritis & Rheumatology 2019, 71, 2100-2111, 10.1002/art.41068.
- Limaye, V.S.; Blumbergs, P.; Roberts-Thomson, P.J.; Idiopathic inflammatory myopathies. Intern. Med. J. 2009, 39, 179–190.
- Harsha Gunawardena; Zoe E. Betteridge; Neil J. McHugh; Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology 2009, 48, 607-612, 10.1093/rheumatology/kep078.
- Vito Racanelli; Marcella Prete; Gerta Musaraj; Franco Dammacco; Federico Perosa; Autoantibodies to intracellular antigens: Generation and pathogenetic role. Autoimmunity Reviews 2011, 10, 503-508, 10.1016/j.autrev.2011.03.001.
- Tomeka L Suber; Livia Casciola-Rosen; Antony Rosen; Mechanisms of Disease: autoantigens as clues to the pathogenesis of myositis. Nature Clinical Practice Rheumatology 2008, 4, 201-209, 10.1038/ncprheum0760.
- Hanbo Yang; Qing-Lin Peng; Liguo Yin; Shanshan Li; Jingli Shi; Yamei Zhang; Xin Lu; XiaoMing Shu; Sigong Zhang; Guo-Chun Wang; et al. Identification of multiple cancer-associated myositis-specific autoantibodies in idiopathic inflammatory myopathies: a large longitudinal cohort study.. Arthritis Research & Therapy 2017, 19, 259, 10.1186/s13075-017-1469-8.
- John C. Hall; Livia Casciola-Rosen; Lesly-Anne Samedy; Jessie Werner; Kristie Owoyemi; Sonye K. Danoff; Lisa Christopher-Stine; Anti-melanoma differentiation-associated protein 5-associated dermatomyositis: expanding the clinical spectrum.. Arthritis & Rheumatism 2013, 65, 1307-1315, 10.1002/acr.21992.
- David Fiorentino; Livia Casciola-Rosen; Autoantibodies to transcription intermediary factor 1 in dermatomyositis shed insight into the cancer-myositis connection. Arthritis & Rheumatism 2012, 64, 346-349, 10.1002/art.33402.
- Takahisa Gono; Shinji Sato; Yasushi Kawaguchi; Masataka Kuwana; Masanori Hanaoka; Yasuhiro Katsumata; Kae Takagi; Sayumi Baba; Yuko Okamoto; Yuko Ota; et al.Hisashi Yamanaka Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology 2012, 51, 1563-1570, 10.1093/rheumatology/kes102.
- David Fiorentino; Lorinda S. Chung; Lisa Christopher-Stine; Lisa Zaba; Shufeng Li; Andrew L. Mammen; Antony Rosen; Livia Casciola-Rosen; Most Patients With Cancer-Associated Dermatomyositis Have Antibodies to Nuclear Matrix Protein NXP-2 or Transcription Intermediary Factor 1γ. Arthritis & Rheumatism 2013, 65, 2954-2962, 10.1002/art.38093.
- Jan Leipe; Xavier Mariette; Management of rheumatic complications of ICI therapy: a rheumatology viewpoint.. Rheumatology 2019, 58, vii49-vii58, 10.1093/rheumatology/kez360.
- Hiroko Kadota; Takahisa Gono; Yuichiro Shirai; Yuka Okazaki; Mitsuhiro Takeno; Masataka Kuwana; Immune Checkpoint Inhibitor-Induced Myositis: a Case Report and Literature Review. Current Rheumatology Reports 2019, 21, 10, 10.1007/s11926-019-0811-3.

