 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | A.S.N.. Reddy | + 6731 word(s) | 6731 | 2021-03-16 03:42:40 | | | |
| 2 | Bruce Ren | Meta information modification | 6731 | 2021-03-19 02:17:58 | | |
Video Upload Options
RNAs transmit information from DNA to encode proteins that perform all cellular processes and regulate gene expression in multiple ways. From the time of synthesis to degradation, RNA molecules are associated with proteins called RNA-binding proteins (RBPs). The RBPs play diverse roles in many aspects of gene expression including pre-mRNA processing and post-transcriptional and translational regulation. In the last decade, the application of modern techniques to identify RNA–protein interactions with individual proteins, RNAs, and the whole transcriptome has led to the discovery of a hidden landscape of these interactions in plants. Global approaches such as RNA interactome capture (RIC) to identify proteins that bind protein-coding transcripts have led to the identification of close to 2000 putative RBPs in plants.
1. Introduction
DNA, the genetic blueprint of all organisms, controls all life processes through intermediate RNA molecules that dictate the types and levels of proteins made in cells. From the biogenesis to degradation of RNA molecules they are associated with many proteins. RNA–protein interactions are numerous, widespread, and play diverse biologically important roles in all organisms in many processes associated with gene regulation, including generation of coding and non-coding RNAs, transport, translation, and decay of RNAs, and control of diverse processes associated with development and disease. The proteins that interact with RNAs are collectively referred to as RNA-binding proteins (RBPs), a diverse class of proteins characterized by the presence of one or more RNA binding domains, usually alongside other catalytic or functional domains. Over 1800 candidate RBPs have been identified in plants, with over 800 enriched as RBPs in Arabidopsis [1]. Plant RBPs play diverse roles in growth, development, genome organization, stress response, immunity, mRNA processing, and post-transcriptional regulation [1][2][3][4][5][6][7]
RBPs rely on their RNA-binding domains to carry out their biological functions. Several of these classes of domains have been characterized, most notably the RNA-Recognition Motif (RRM), DEAD-box helicases, zinc finger domains, the K homology (KH) domain, the glycine-rich domains, the pentatricopeptide repeats (PPRs), and pumilio/fem-3 binding factors (PUFs) [1][7]. The RNA-binding domains allow RBPs to regulate many different processes: pre-mRNA splicing, pri-miRNA and pre-miRNA processing, polyadenylation, nuclear export, RNA stability, translation, RNA editing, etc. [2]. Moreover, many proteins identified as candidate RBPs lack classical RNA-binding domains, and there is even a high prevalence of metabolic enzymes identified as the RNA-interacting proteins, underscoring the complexity of RNA–protein interactions and the current gaps in understanding [8]. This is in accordance with results from mammalian mRNA-interactome studies, which revealed 23 distinct metabolic enzymes as RBPs [9]. Thus, it has been hypothesized that the moonlighting of metabolic enzymes as RBPs forms a regulatory link between cellular metabolism and RNA fate, known as the RNA-enzyme-metabolite (REM) hypothesis [9][10].
2. Overview of Methods to Detect RNA-Protein Interactions and Their Application to Plants
RNA-binding proteins have become a target of great interest in recent years, and many new methodologies have been developed to analyze the RNA–protein interactome. However, in plants, most research done in this field before ~10 years ago relied entirely on the use of indirect or in vitro methods to identify RNA and protein interaction, such as gel shift assay, mutant and knockout screening, nucleic acid-binding assay, and other classical genetics and cell biological techniques [2][11][12][13]. These techniques have contributed significantly to understanding the functions of RBPs in plant biology (see Section 3) but have since been superseded by the development of high throughput and global methods to analyze RNA and protein interactions. These new techniques were developed first in mammalian systems and a few have been used increasingly in plants. Below, we briefly describe these methods and their limitations, especially with respect to applying them in plants.
These techniques fall into three categories: (i) approaches that focus on identifying RNA targets of a candidate RBP, i.e., protein-to-RNA, (ii) approaches that focus on identifying the proteins interacting with an RNA of interest, i.e., RNA-to-protein, and (iii) global approaches (Figure 1). The vast majority of work that has been done in this field in plants has focused on the interacting partners of a single RNA or protein of interest (the bait), but recently the development of RNA-interactome capture (RIC) and its application to plants has allowed a global view of the plant RBPome.
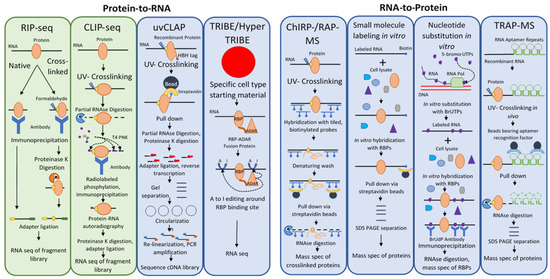
2.1. Methods That Use a Protein Bait to Identify Its RNA Targets (Protein-to-RNA) and RBPs Identified Using These Methods in Plants
Among the first techniques developed to identify direct targets of RBPs in vivo was RNA immunoprecipitation or RIP [15][16] (Table 1). The basic idea of the RIP approach is simple and involves the use of an antibody against a protein of interest (Figure 1, RIP-seq). The lysate of cells expressing the protein of interest is incubated with antibody immobilized on beads, which are then washed and the proteins on the beads digested. The pool of RNA remaining is used to identify putative binding RNA targets. With the development of high throughput sequencing technologies, methodologies that used such sequencing platforms became known as RIP-seq [17].
| Method | Pros | Cons | Plant Refs |
|---|---|---|---|
| Protein-to-RNA | |||
| RIP-seq | No genetic trans., reversible crosslinking, well established in plants, no radiolabeling | Uses antibody–antigen interaction, non-specific crosslinking, large amounts of starting material, no info on RBP site | He et al., 2009, Streitner et al., 2012; Yin et al., 2012; Rowley et al., 2013; Bardou et al., 2014; Francisco-Mangilet et al., 2015; Xing et al., 2015; Bazin et al., 2018; Marmisolle et al., 2018; Schmid et al., 2019; Tian et al., 2019 |
| CLIP-seq | No genetic trans., provides info. on the RBP binding site, well-established in plants | Uses antibody–antigen interaction, uses radiolabeling, large amounts of starting material | Meyer et al., 2017; Zhang et al., 2015 |
| HITS-CLIP | Increased coverage | As CLIP-seq | Zhang et al., 2015 |
| PAR-CLIP | More efficient UV-crosslinking | As CLIP-seq, favors certain RBP-RNA interactions | None |
| iCLIP | Increases precision of RBP site prediction | As CLIP-seq | Meyer et al., 2017 |
| dCLIP | Permits comparisons across all CLIP exps. | As CLIP-seq | None |
| uvCLAP | Tight binding affinity, uniform pulldown efficiency, quantify background, no radiolabeling, no antibodies | Not in plants, needs genetic trans., may alter RNA–protein interactions, no info on RBP site, large amounts of starting material | None |
| TRIBE/HyperTRIBE | No pull down, small amounts of starting material, no radiolabeling, no antibodies | Not in plants, needs genetic trans., editing occurs in a wide range around the binding site, no info on RBP site | None |
| RNA-to-Protein | |||
| ChIRP-MS/RAP-MS | High affinity interaction, no genetic trans., no radiolabeling, no antibodies | Not in plants, no info on RBP site, large amounts of starting material | None |
| RNA Small Molecule Labeling | No genetic trans., no radiolabeling, no antibodies | In vitro only | None |
| RNA Nucleotide Substition | No genetic trans., no radiolabeling, no antibodies | In vitro only | None |
| RNA Aptamer Pulldown | High affinity interaction, many aptamers, no radiolabeling, no antibodies | Not in plants, needs genetic trans., no info on RBP site, may alter RNA–protein interactions, large amounts of starting material, may be prone to aggregation | None |
RIP can also involve RNA–protein crosslinking, creating covalent bonds between the protein and its RNA ligands. Reversible crosslinking is accomplished using formaldehyde and reversed via heat treatment [18]. The drawbacks of this approach are that the specificity of the results depends on the strength of the antibody–protein interaction, and that formaldehyde treatment also catalyzes DNA-protein and protein–protein crosslinking, leading to the identification of indirect as well as direct targets of an RBP.
Crosslinking and immunoprecipitation (CLIP) builds on RIP by replacing formaldehyde crosslinking with UV-crosslinking to covalently link proteins with RNA molecules within several angstroms distance (i.e., bound by the protein) (Table 1). The RNA–protein complexes are selected after cell lysis using immunoprecipitation [19]. Partial digestion of the bound RNA allows a rough approximation of the binding site, followed by phosphorylation of the complexes with radio-isotope. The covalently bound RNA–protein complex is then rigorously washed, separated via SDS-PAGE, and transferred to a nitrocellulose membrane. The protein is then removed using proteinase K, linkers are ligated to the collected RNA fragments, and the fragment library is cloned after reverse transcription and then sequenced (Figure 1, CLIP-seq). There are many derivative techniques based on the basic CLIP-Seq principle. High-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP) in place of traditional sequencing, which placed a limitation on the richness of data that could be generated by a CLIP-Seq experiment, allows more data to be extracted from CLIP fragment libraries (Table 1). This allowed the identification of over 1000-fold more unique binding sites compared to CLIP with traditional sequencing techniques, although this leaves one with the opposite problem—a plethora of data to sift through and discern signal from noise [20]. CLIP-based methods were further improved with the advent of CLIP experiments using photoactivatable ribonucleosides (PAR) to enhance the efficiency of crosslinking. PAR-CLIP incorporates 4-thiouridine into transcripts in vivo, which forms covalent bonds with interacting proteins under UV far more efficiently than random UV RNA–protein crosslinking; the approach improved RNA recovery 100- to 1000-fold [21].
Thus far, CLIP techniques were limited by the fact that reverse transcriptase often terminates prematurely when met with a residual amino acid covalently bound to a nucleotide at a crosslinking site causing such reads to be lost during standard CLIP library preparation. Individual-nucleotide resolution CLIP (iCLIP) was developed to compensate for this problem [22]. iCLIP captures truncated cDNAs using a cDNA self-circularization step in place of the previously used inefficient RNA ligation step in library preparation [22]. CLIP experiments also suffered from high experimental failure rates due to their technical complexity, and enhanced CLIP (eCLIP) was developed to address these issues. eCLIP decreases the amount of amplification necessary and uses random-mer barcode adapters ligated at the termination site of reverse transcriptase (the UV crosslinked nucleotide) to maintain analysis of RBP binding sites. Furthermore, the protocol omits the radiolabeling step and uses a size-matched control without immunoprecipitation to eliminate non-specific RNA interactions from the datasets [23].
The CLIP technique was also simplified by the development of simplified CLIP (sCLIP), which avoids radiolabeling by biotinylating the RNA for visualization, and uses polyadenylation and random-mer barcoding to uniquely identify RNAs and reduce the requirement for PCR amplification [24]. Another technique designed to avoid the use of radiolabeling, termed irCLIP for its use of an infrared dye, also used biotin labeling of the RNA—a biotinylated and infrared dye-conjugated 3’ adapter was ligated to the RNA, allowing visualization of RNA–protein complexes without autoradiography [25]. irCLIP allows the use of 250 times less starting material compared to iCLIP, and although comparisons were not performed with eCLIP or sCLIP, it seems likely that irCLIP lowers the starting material requirement most significantly.
With the advent of HITS-CLIP, many computational tools were developed in order to handle the large datasets produced by HITS-CLIP experiments. One of the most widely used of these is known as dCLIP, a program created to allow comparison of differential binding in different CLIP experiments [26]. dCLIP normalizes CLIP-seq data from different experiments using an application of a Bland-Altmann plot called an MA plot, then uses a Hidden Markov Model to detect shared or distinct binding sites across experiments. dCLIP has the advantage of being a universal computational tool for all types of CLIP-seq experiments; HITS-CLIP, PAR-CLIP, and iCLIP, and to allow comparison among them [26].
CLIP-Seq and its derivatives are powerful techniques but have significant limitations. Namely, CLIP (and its derivative methodologies) are all limited by their reliance on the antibody–antigen interaction; this limits the stringency of washing conditions to those that will not disrupt the antibody–antigen interaction [27]. Thus, the acquisition of a high-affinity antibody is critical for such experiments, and generally cannot be guaranteed. Even meaningful CLIP experiments contain significant noise in the form of proteins that were not eluted under the weak washing conditions, or in the form of proteins that co-immunoprecipitated [28]. Furthermore, CLIP relies on radiolabeling of bound RNA, a prohibitive procedure due to its cost, difficulty, and health hazards [29]. Finally, because of the low efficiency of CLIP techniques, they require large amounts of starting material, on the order of thousands of cells. This means that studies of RNA–protein complexes in specific cell types (which cannot be amassed in the thousands) are forced to use starting material of a mixed population of cell types, lowering the signal to noise ratio in their results [30]. Other techniques, discussed below, have been developed to avoid these limitations.
UV-crosslinking and affinity purification (uvCLAP) was developed as a radiolabeling- and immunoprecipitation-free alternative to CLIP methodologies [29] (Figure 1, uvCLAP). Instead of using an antibody–antigen interaction, uvCLAP relies on the tight interaction of the His6-biotinylation sequence-His6 (HBH) tag with beads that bind polyhistidine-tagged proteins, and then with the even more stringent interaction with streptavidin beads. The RNA is partially digested with RNAseI and the RNA ends are repaired with T4 polynucleotide kinase. Adapters are then ligated to the RNA fragments and reverse transcribed with barcoded primers. The cDNA products are then separated on a polyacrylamide gel, circularized to capture truncated cDNA products (as in iCLIP), linearized, and amplified with PCR.
The use of tandem affinity purification in this approach allows confidence that pulldown efficiency will be similar across conditions, experiments, and laboratories, in comparison with immunoprecipitation approaches in which every antibody–antigen interaction has a unique affinity [29]. uvCLAP also allows the quantification of nonspecific background noise, increasing its specificity. The drawback of this approach is the need for a genetic transformation with an HBH-fused construct prior to affinity purification. Although it is relatively unlikely when done carefully, such transformations could potentially alter RNA–protein interactions from their natural state. Moreover, this introduces extra steps for each RNA-binding protein studied; the significance of this drawback will depend entirely on the ease of genetic transformation in the model system being used.
The TRIBE (targets of RNA-binding proteins identified by editing) and HyperTRIBE approaches were developed in response to the severe limitations of CLIP-based techniques in identifying cell type-specific RNA–protein interactions [30]. TRIBE was developed first; it uses the RNA-editing enzyme ADAR’s (adenosine deaminase acting on RNA) to convert adenosines to guanines, leaving telltale signals in edited RNA (Figure 1, TRIBE/HyperTRIBE). In this approach, ADAR’s double-stranded RNA-binding motifs are replaced with the sequence of an RNA-binding protein of interest to create a fusion protein that targets ADAR’s RNA-editing activity to the RNA targets of the fused RBP. The RNA is sequenced, and detection of editing events indicates the binding of the fusion protein, and thus the RBP of interest.
The original TRIBE technique had the opposite problem as most CLIP experiments: it identified only about 25% of the target RNAs identified by CLIP techniques for the same RBP, and is thought to have had a false negative problem, rather than CLIP’s false positive problem. It was found that ADAR’s editing rate was low due to a sequence specificity for UAG and a double-stranded structure surrounding the edited adenosine [31].
To compensate for these weaknesses, hyperTRIBE was developed by introducing the E448Q mutation in ADAR, which lowers ADAR’s sequence and structure preferences and increases editing efficiency [31]. This mutation increased the number of detected editing events by over 20 times, while increasing the number of detected edited transcripts by 8 times. HyperTRIBE is able to identify about two-thirds of CLIP-identified target RNAs.
This approach has the advantages of avoiding the use of immunoprecipitation and radiolabeling, requiring only a small amount of starting material, and being simple. Like uvCLAP however, it also requires genetic transformation, and in comparison to both uvCLAP and CLIP techniques, has the drawback of providing no information as to the specific binding site on the RNA (as ADAR edits sites within up to 500 nucleotides of known CLIP sites). CLIP remains the method of choice if information about an RBP’s binding site on an RNA is desired, whereas HyperTRIBE is desirable if interested in RNA–protein complexes in specific cell types or if only small amounts of starting material are available [31].
Among the methods described above, the only protein-to-RNA techniques that have been used in plants to-date are RIP-seq and CLIP-seq. As discussed below, the application of these techniques to several RBPs has revealed their role in several processes.
2.1.1. Regulation of RNA Processing
RIP-seq was used to demonstrate that the Arabidopsis Serine- Arginine-rich (SR) protein SR45 directly or indirectly associates with over 4000 RNAs in vivo, regulating constitutive and alternative splicing, post-splicing processing of 30% of ABA signaling genes, and over 300 intron-less RNAs [32] (Table 2). This indicates that SR45 exerts multimodal influence over mRNA processing, differentially regulating intron-containing and intron-less RNAs. The action of SR45 is defined by cis-elements in its RNA targets; four motifs were identified, two of which bear the hallmarks of exonic splicing regulators and two which showed peaks in the intronic regions of 5’ and 3’ splice sites. One of these motifs (M1; GAAGAA) was also found to be enriched in SR45’s intron-less targets [32]. Another study found 1812 RNAs associated with SR45, 81 of which were subject to alternative splicing mediated by the GGNGG motif in both activation and repression of splicing events [33]. These results further define SR45 as a splicing regulator whose activity cannot be easily defined as a positive or negative regulator, possibly explained by the fact that SR45 itself is alternatively spliced and its splice isoforms display differential expression. SR45 produces two splice isoforms, SR45.1 (long) and SR45.2 (short), the long isoform acting as a positive regulator in the salt stress response in Arabidopsis [34]. In rice, SR45 is stabilized through interactions with an immunophilin (OsFKBP20-1b), which plays an essential role in a positive regulation of transcription and splicing of stress response genes during abiotic stress [35]. THO2, a member of the Transcription-Export (THO/TREX) complex, was shown via RIP to participate in the generation of microRNAs; THO2 mutants showed both a decrease of miRNA accumulation and alterations in the splicing patterns of SR proteins, suggesting that the THO/TREX complex plays a role in alternative splicing [36].
| RBP | Plant System | Method | Number of RNA Targets | References |
|---|---|---|---|---|
| AGO4 | Arabidopsis thaliana | RIP | 2 | Wierzbicki et al., 2009 |
| AtGRP7 | Arabidopsis thaliana | RIP-seq/iCLIP | 452/858 | Streitner et al., 2012; Meyer et al., 2017 |
| AtNSRa | Arabidopsis thaliana | RIP-seq | >2000 | Bardou et al., 2014; Bazin et al., 2018 |
| AtNSRb | Arabidopsis thaliana | RIP-seq | >2000 | Bardou et al., 2014; Bazin et al., 2018 |
| CPsV 24K (viral) | Nicotiana benthamiana | RIP | 2 | Marmisolle et al., 2018 |
| CPsV 24K (viral) | Nicotiana benthamiana | RIP | 2 | Marmisolle et al., 2018 |
| CSP1 | Arabidopsis thaliana | RIP-chip | >6000 | Juntawong et al., 2013 |
| IDN2 | Arabidopsis thaliana | RIP | 1 | Zhu et al., 2013 |
| FCA | Arabidopsis thaliana | RIP | 1 | Tian et al., 2019 |
| HLP1 | Arabidopsis thaliana | HITS-CLIP | >5000 | Zhang et al., 2015 |
| KTF1 | Arabidopsis thaliana | RIP | 1 | He et al., 2009 |
| NSF | Oryza sativa | RIP | ? | Tian et al., 2020 |
| PUMPKIN | Arabidopsis thaliana | RIP-seq | 5 | Schmid et al., 2019 |
| PDM1 | Arabidopsis thaliana | RIP | 1 | Yin et al., 2012 |
| Rab5a | Oryza sativa | RIP | ? | Tian et al., 2020 |
| RBP-L | Oryza sativa | RIP | ? | Tian et al., 2020 |
| RBP-P | Oryza sativa | RIP | ? | Tian et al., 2020 |
| SR45 | Arabidopsis thaliana | RIP-seq | >4000/>1800 | Xing et al., 2015; Zhang et al., 2017 |
| THO2 | Arabidopsis thaliana | RIP | 6 | Francisco-Mangilet et al., 2015 |
RIP was used to show that the glycine-rich RBP AtGRP7 modulates alternative splicing in Arabidopsis [37]. A later study using both RIP-seq and iCLIP found 452 (RIP-seq) and 858 (iCLIP) RNA targets of AtGRP7 [38]. AtGRP7 alters the circadian regulation of its targets and seems to act in both alternative splicing and alternative polyadenylation (APA) [38] (Table 2).
Nuclear speckle RNA binding proteins (NSRs) have also been shown via RIP-seq to regulate mRNA processing, alternative splicing, and long noncoding RNA (lncRNA) prevalence [39] (Table 2). An NSR and an alternative splicing competitor (ASCO) lncRNA were shown to form a regulatory module of alternative splicing, in which the ASCO displaces an alternative splicing target from an NSR complex to modulate alternative splicing during development [39]. NSRs affected alternative splicing of hundreds of genes in Arabidopsis, and RIP-seq of an NSRa fusion protein showed that lncRNAs are also targets of NSRs, likely modulating their alternative polyadenylation or splicing as observed with the COOLAIR lncRNA to regulate cross-talk between auxin and immune response [40].
HITS-CLIP was used to identify genome-wide targets of HLP1, an hnRNP A/B protein that binds preferentially to A- and U-rich elements around cleavage and polyadenylation sites of transcripts involved in RNA metabolism and flowering to target APA [41] (Table 2). HLP1 suppresses Flowering Locus C (FLC) to release repression of flowering in Arabidopsis and control reproductive timing [41]. NSR knockout mutants showed modified APA and differential expression of the lncRNAs COOLAIR, produced from antisense transcripts generated from FLC, and function in the release of repression of flowering through suppression of FLC [40].
Using RIP-seq, the pentatricopeptide repeat protein PDM1 was shown to mediate cleavage of a transcript from polycistronic to monocistronic fragments in chloroplasts of Arabidopsis [42] (Table 2).
2.1.2. Trafficking and Translocation
In rice, RIP-seq was used to show that RNA-binding protein-P (RBP-P) is an RNA-binding protein that plays a role in endosomal trafficking of glutelin and prolamine mRNAs, working to anchor the RBP-bound mRNAs to the endosome via the quaternary complex and transport it to the ER Subdomain for translation, coopting endosomal trafficking [43] (Table 2). RBP-L, an interacting partner of RBP-P, likely plays a coordinating role in subcellular trafficking of its mRNA targets, mediated by its 3’ UTR [44] (Table 2).
2.1.3. Chaperoning
In Arabidopsis, a unique combination of RIP and microarray approaches (RIP-Chip) was used to demonstrate that the cold shock protein 1 (CSP1) acts as an RNA chaperone of polysomes to improve the translation of RNA targets at low temperatures [45] (Table 2).
2.1.4. Gene Silencing
The RNA-directed DNA methylation effector KTF1 was identified via RIP as an RBP that binds Pol V scaffold transcripts to recruit argonaute 4 (AGO4) and its siRNAs for chromatin remodeling-mediated gene silencing [15] (Table 2). AGO4 and RNA polymerase V cooperate with 24 nt siRNAs in this process; siRNAs bound to AGO4 guide AGO4 to target loci through complementary base-pairing with nascent Pol V transcripts, where AGO4 recruits DNA modification factors such as DNA methyl-transferase DRM2 to methylate the chromatin and thus silence the affected genes [15][45] Based on RIP observations that the protein INVOLVED IN DE NOVO 2 (IDN2) is a lncRNA-binding protein that interacts with the SWItch/Sucrose Non-Fermentable (SWI/SNF) nucleosome remodeling complex, lncRNAs are thought to base-pair with siRNAs bound by AGO4 to position the SWI/SNF complex and thus target nucleosome remodeling, leading to decreased transcription by Pol II [46][47] (Table 2). RIP was also shown to be usable in Arabidopsis for the detection of lncRNAs generated by specialized polymerases [48].
2.1.5. Viral RNA Suppression
A modified RIP-seq assay was developed for the detection of RNAs of heterologous origin in plants and applied to transiently expressed nuclear epitope-containing proteins in Nicotiana benthamiana, but to-date this method has not been used for its intended purpose of detecting viral RNAs in plant cells [49].
2.1.6. Other RBPs
The plastid UMP kinase (PUMPKIN) has been shown via RIP-seq to associate with several RNAs in vivo, altering their metabolism thereby [50] (Table 2). This suggests that while PUMPKIN is primarily a metabolic enzyme, it may have a moonlighting function as an RBP, potentially for the purpose of coupling RNA and pyrimidine metabolism [50].
2.1.7. Perspective on the Application of Protein-to-RNA Methods in Plants
Despite the breadth of techniques available for use in elucidating RNA–protein interactions in vivo, CLIP and its derivatives remain the most tenable non-global approach for use in plants. RIP has also been used extensively and is suitable for certain experimental purposes. There still remain several techniques used in other organisms to probe interactions between a protein of interest and RNAs that have yet to be successfully adapted, or even tried, in plants. These are opportunities for advancement in plant RNA biology, but if adapted into plants should be modified to include the best features and optimizations of the already-proven RIP and CLIP approaches.
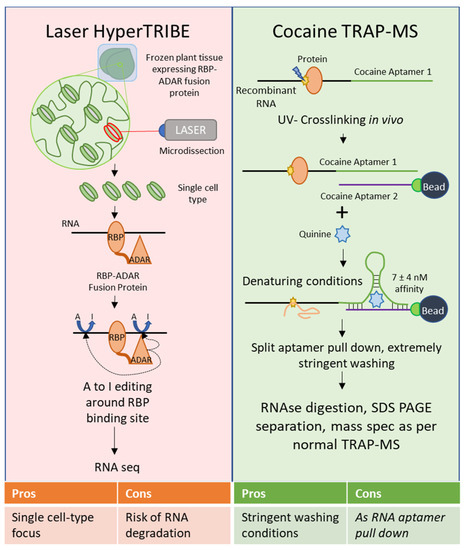
Several of these techniques show particular promise; TRIBE, and particularly HyperTRIBE, have not been used in plants as yet, but if viable would overcome the signal to noise ratio problems inherent in CLIP. HyperTRIBE outperforms CLIP when using a small amount of starting material, such as a few cells of homogenous origin. Unfortunately, techniques used to select cells of a single type from a heterogeneous sample in mammalian systems, such as flow cytometry, are not tenable in plants without significantly altering the cell state (i.e., generating protoplasts by degrading the cell wall) [51]. Laser microdissection of plant tissues seems the most viable route for selecting cells of a particular type in plants, and HyperTRIBE would allow the use of smaller amounts of starting material than were previously used for RNA-Seq after laser microdissection [52]. Focus on single cell-types is a necessary next step for plant biology to throw off the albatross of whole-plant and tissue heterogeneity, and HyperTRIBE combined with laser microdissection would represent progress toward that goal in the field of RBPomics (Figure 2). However, laser microdissection requires a somewhat more extended time between sample harvesting and freezing due to the fixation step, which could result in increased RNA degradation after harvesting. Even so, transcriptional profiling has been performed successfully using cells harvested via this technique [52].
2.2. Methods That Use an RNA Bait to Identify Binding Proteins (RNA-to-Protein)
RNA antisense purification mass spectrometry (RAP-MS) is a technique used to purify long noncoding RNAs and their interacting proteins with complementary, tiled, biotinylated DNA probes bound to magnetic streptavidin beads [53] (Figure 1, ChIRP-/RAP-MS). RAP-MS starts with UV crosslinking of RNA to interacting proteins in vivo. The crosslinked RNA–protein complexes are then extracted under denaturing conditions to disrupt non-covalent interactions, and the complexes are hybridized with ~120 nt biotinylated DNA probes bound to magnetic beads. After washing, the RNA is digested, and the protein pool is analyzed using mass spectrometry (MS). This method also uses stable isotope labeling by amino acids in culture (SILAC) to label proteins, allowing quantitative comparisons to be made with mass spectrometry [54].
Comprehensive identification of RNA-binding proteins by mass spectrometry (ChIRP-MS) is a related technique predating RAP-MS by several years [55]. It also uses tiled biotinylated DNA probes bound to magnetic streptavidin beads and RNA–protein crosslinking, although the probes used were only 20 nt in length and formaldehyde crosslinking was chosen instead of UV crosslinking. The use of formaldehyde crosslinking has the advantage of being reversible, and ChIRP-MS studies are able to reverse crosslinking while keeping both protein and RNA components intact and allowing further analyses on both [56]. However, formaldehyde crosslinking also catalyzes the crosslinking of protein–protein and protein-DNA interactions.
The technique known as PIP-Seq has been used successfully to elucidate important RNA–protein interactions governing the differentiation of root hair cells [57]. PIP-seq identifies RNA–protein interactions with precise RNA binding sites when paired with a technique capable of identifying individual interacting RBPs. PIP-seq uses formaldehyde crosslinking to covalently bond RNA to interacting proteins, followed by high-throughput sequencing. The sample is split into a matrix of four: one sample with RBPs intact treated with single-stranded RNA nuclease (ssRNAse), one without RBPs treated with ssRNAse, one with RBPs treated with double-stranded RNA nuclease (dsRNAse), and one without RBPs treated with dsRNAse. The use of ss- and dsRNAse in the presence and absence of binding RBPs allows both RNA structure and RBP protection (and thus binding) to be predicted [57].
Recently, a CRISPR-based system called CRUIS (CRISPR-based RNA-United Interaction System) was developed in mammals [58]. CRUIS uses transient expression to couple the RNA-tracking capabilities of dCas13a with a fused proximity protein, Pafa, which labels surrounding RNA-binding proteins. These labeled proteins can then be identified via mass spectrometry. CRUIS was shown to be roughly as efficient as CLIP and identified novel protein targets [58]. The advantage of this technique is that it captures truly in vivo interactions without the potential for spurious interactions to form during lysis and wash steps, but it remains to be seen whether it has a false positive problem. The Pafa proximity labeling protein lacks the specificity of UV crosslinking for angstrom-level RNA–protein interactions, potentially leading to the labeling of indirectly interacting proteins.
There are a number of RNA to protein methods that are useful for in vitro studies but are not applicable to in vivo work. Among these is the labeling of RNA with small molecules [59]. In this RNA to protein approach, small molecules are covalently bonded to an RNA of interest in vitro, then incubated with cell lysate and pulled down using an immobilized receptor for the small molecule ligand (Figure 1, small molecule labeling in vitro). Common forms of this technique include biotin labeling, desthiobiotin labeling, and digoxigenin labeling. Unfortunately, because of the chemical reactions necessary to label an RNA of interest, small molecule RNA labeling is usually not appropriate for in vivo studies.
Another exclusively in vitro approach is nucleotide substitution in RNA [59]. Here, RNA is transcribed in vitro in the presence of a heavy metal-modified dNTP, incorporating the modified nucleotide into the transcript. Immunoprecipitation can then be carried out using an antibody against the modified nucleotide (Figure 1, nucleotide substitution in vitro). The drawback of this approach is that the charge of the heavy metal-modified nucleotide can strongly affect the charge distribution, structure, and protein binding of the RNA of interest.
Whereas the uvCLAP approach uses modifications to the protein primary structure, RNA aptamer pulldown (also known as tandem repeat affinity purification mass spectrometry, or TRAP-MS) uses modifications to the RNA primary and secondary structures, followed by tandem affinity purification [59]. RNA aptamers are short oligonucleotide sequences that reliably assume a secondary structure under physiological conditions, which tightly interacts with a target molecule—the ligand. The affinities of these interactions can be equivalent to or greater than those of antibody–antigen interactions [60][61][62][63]. An RNA aptamer is introduced into an RNA of interest either in vitro or in vivo, the lysate is passed over a column containing immobilized ligand, washed, and ribonucleoprotein complexes are eluted. Interacting proteins are identified via mass spectrometry. This, like RAP-MS/ChIRP-MS, is one of the few in vivo methods to identify ribonucleoprotein complexes in the RNA-to-protein direction.
There are many well-studied RNA aptamers used for such studies; some of the most commonly used are the PP7, S1, D8, tobramycin, streptomycin, Csy4 (H29A), Mango, and MS2 aptamers [59][60][61][62]. Only the MS2 aptamer will be discussed in detail here. This aptamer exploits the tight, highly specific interaction between the coat protein (MCP) of the bacteriophage MS2 and a 19nt RNA hairpin structure from the bacteriophage’s genome, which the virus presents on the surface of its genome to assemble its coat protein [60]. Repeats of the MS2 hairpin structure are inserted at the 3’ end of an RNA of interest, while a fusion protein of MCP and maltose-binding protein (MBP) is immobilized on amylose beads. After pulldown, the protein-RNA-MCP-MBP complex is eluted using excess maltose, which MBP binds preferentially (Figure 1, TRAP-MS). RNA aptamer pulldown has the disadvantage of requiring genetic transformation, which may alter the structure of the RNA of interest and thus distort the pool of RNA binding proteins associated with it. Furthermore, the presence of the RNA aptamer may risk aggregation.
Two other RNA-to-protein techniques were developed in the last year in non-plant systems. One of these methods targets engineered peroxidase (APEX) with MS2 or Cas13 to a specific RNA. APEX targeting uses either the MS2-MCP interaction or an engineered CRISPR-Cas13 interaction to target the biotinylation activity of APEX2 to proteins proximal to target RNAs in vivo [64]. After rapid, one-minute biotin labeling, cells are lysed and pulled down using streptavidin beads. Isolated proteins are identified using liquid chromatography-mass spectrometry (LC-MS). This method was based on the RNA proximity biotinylation (RNA-BioID) and APEX RNA immunoprecipitation (APEX-RIP) approaches. RNA-BioID uses MCP to target a biotin ligase (BirA*) to an MS2-tagged RNA of interest [65]. APEX-RIP uses the promiscuous engineered peroxidase APEX2 expressed by live cells to target cellular components of interest and biotinylate proximal proteins during a short pulse of treatment with hydrogen peroxide and biotin-phenol [66]. Following biotinylation, labeled proteins are crosslinked to proximal RNAs using formaldehyde and pulled down using streptavidin beads, along with co-eluting RNAs. APEX targeting improves on BioID by decreasing the amount of time necessary for biotin labeling [66]. Although it is claimed [66] that APEX2 does not label distal proteins due to the short half-life of the biotin-phenoxyl radical it generates, it is unknown whether APEX2 may label proteins interacting indirectly with the target RNA. Compared to crosslinking, which establishes a hard limit on the distance of RNA–protein interaction, this may raise a concern of false positives when using APEX targeting.
The second method is called CRISPR-assisted RNA–protein interaction detection (CARPID). This method was also inspired by APEX-based approaches but uses the engineered biotin ligase BASU instead of APEX2 [67]. Using a nuclease-activity-free RNA targeting dCasRx to tether BASU to RNAs of interest, CARPID labels interacting proteins via biotinylation, followed by pull-down with streptavidin beads [67]. This method was able to identify RBPs interacting with lncRNAs but requires a longer labeling period as compared to APEX targeting.
Perspective on the Use of RNA-to-Protein in Plants
There is much room for improvement in the RNA-to-protein direction, particularly considering that none of these techniques have been used in plants to-date. ChIRP-MS in particular would be an attractive technique to attempt in plants for the following reasons: it avoids the need for antibody generation used in RIP and CLIP, it does not use radiolabeling, it permits denaturing conditions and stringent washes, and it does not require genetic transformation. However, as previously described it cannot provide any information regarding the binding site of an RBP.
RNA aptamer-mediated pull-down techniques could also be an area for advancement. These approaches do require genetic transformation and could potentially result in altered RNA secondary structure (depending on the aptamer used), but their potential to exceed the antibody–antigen affinity limitations and avoid the antibody generation variabilities of CLIP makes them attractive nevertheless. However, because most of the annotated RNA aptamers in use rely on the binding capabilities of partner proteins (such as the MS2 stem loop’s binding by the MS2 viral coat protein MCP), their use limits the stringency of washing conditions; denaturing conditions cannot be used during incubation and washing to prevent the formation of post-lysis ribonucleoprotein complexes without denaturing the aptamer’s binding partner and thereby compromising pull down. Of those that do not rely on protein partners, few match the affinity granted by the polyA-oligo(dT) interactions used in other techniques, such as RNA-interactome capture.
It might be advantageous to develop nucleotide-nucleotide RNA aptamers to increase the binding affinity, such as by applying a split RNA aptamer. Split aptamer approaches involve separating out an existing aptamer, such as an RNA that forms a tight stem-loop secondary structure, into two fragments that tightly interact in the presence of a ligand; thus, one fragment of the aptamer is appended to a transcript of interest, and the second is immobilized on a nonreactive bead. For example, the cocaine aptamer has successfully been split and used as a biosensor [68]. Although its target as a biosensor is cocaine, the split aptamer actually shows 30- to 50-fold greater affinity for quinine over cocaine, binding at an affinity of 7 ± 4 nM [68][69]. Use of the cocaine aptamer in the presence of quinine during pull-down could potentially tolerate extremely stringent washing conditions. For a summary of these suggested techniques, see Figure 2. Proximity biotinylation-based methods, such as APEX targeting and CARPID could theoretically be used in plants and would be of interest due to their status as RNA-to-protein methods with some modifications.
2.3. Global RNA-Protein Interactome
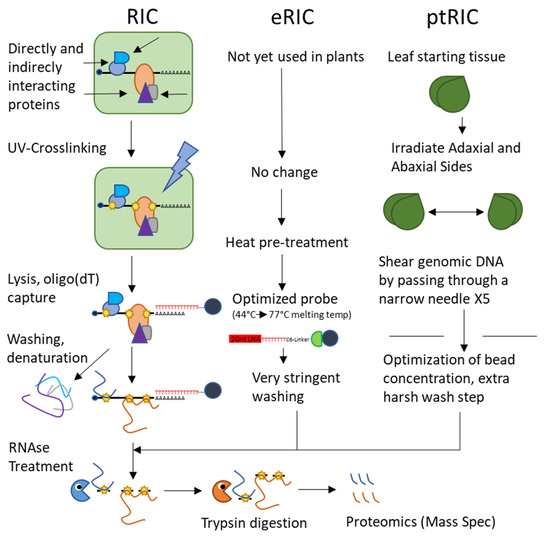
Until very recently, there was only one currently available global approach to capturing the plant RBPome, called RNA-interactome capture (RIC). RIC uses techniques common to directed RNA–protein interaction studies, beginning with the UV-crosslinking of interacting proteins to their partner RNAs as in CLIP and PAR-CLIP. The cell lysate is then passed over oligo(dT)-magnetic beads under denaturing conditions to pull down polyA RNAs and the denatured proteins covalently bonded to them. After stringent washes to elute any non-covalently interacting proteins, the RNA is enzymatically digested and the protein sample is subjected for proteomics via mass spectrometry [70] (Figure 3). This technique is powerful but limited by its restriction to polyA RNA.
The RIC technique was adapted into plants several years ago by a trio of studies using cell suspension cultures, seedling leaves, leaf mesophyll protoplasts, and etiolated whole seedlings [8][71][72]. These studies identified between 300 and 1200 RBPs, all showing enrichment of proteins containing canonical RNA-binding domains. They also all identified a significant proportion of proteins lacking a canonical RNA-binding domain and playing no known role in RNA biology, underscoring how poorly described RNA–protein interactions are in plants. Finally, all three studies found significant proportions of enzymes involved in intermediate metabolism making up the RBPome, suggesting that the RNA-enzyme-metabolite hypothesis may be a valid consideration in plants as well as mammals.
These studies provide us with a perspective on heterogenous plant samples grown under normal conditions and provide a baseline against which future studies may compare the results of experiments using the array of sample types described. Since their publication, the RIC method has been applied to Arabidopsis cell cultures grown under drought stress (using PEG to simulate drought conditions in culture) to identify 150 RBPs responsive to drought stress [73]. Similarly, RIC was used to probe modifications of the spliceosome and its RBPs in response to drought, identifying 44 spliceosomal proteins and 32 proteins associated with stress granules [74]. Like the previous studies, this work identified many metabolic enzymes interacting with RNA, comprising proteins involved in carbohydrate metabolism and the glycolytic and citric acid pathways. Recently, several optimizations of the RIC protocol—deemed enhanced RNA interactome capture or eRIC—were described, but these modifications have yet to be applied to plants [75] (Figure 3). Separately, RIC has been optimized for leaf tissue (termed plant RNA interactome capture or ptRIC) by adjusting UV conditions, irradiating both adaxial and abaxial surfaces of leaves, increasing the stringency of washing conditions, and shearing genomic DNA by passing the RNA-loaded beads through a narrow needle [76] (Figure 3). It remains now for RIC or its derivatives to be used to view the changes of the RBPome in response to biotic and abiotic stresses beyond drought.
Very recently, a new method for the identification of RNA–protein interactions has been adapted from bacterial and mammalian systems, known as orthogonal Organic Phase Separation, or OOPS. This method uses UV-crosslinking, similar to other techniques, and acidic guanidiniumthiocyanate-phenol-chloroform (AGPC) phase separation to collect RBPs at the interface between the aqueous and organic phases [77]. OOPs has the advantage of being simpler than many other techniques and of not requiring mRNA pulldown, thus capturing RBP interactions with all types of RNA rather than solely coding RNAs. OOPS was applied in Arabidopsis to identify 468 RBPs, 232 of which were enzymatic putative RBPs [78].
References
- Marondedze, C. The increasing diversity and complexity of the RNA-binding protein repertoire in plants. Proc. R. Soc. 2020, 287, 20201397.
- Lorkovic, Z. Role of plant RNA-binding proteins in development, stress response and genome organization. Trends Plant Sci. 2009, 14, 229–236.
- Woloshen, V.; Huang, S.; Li, X. RNA-Binding Proteins in Plant Immunity. J. Pathog. 2011, 2011, 278697.
- Huh, S.; Paek, K. Plant RNA binding proteins for control of RNA virus infection. Front. Physiol. 2013, 4, 397.
- Lee, K.; Kang, H. Emerging Roles of RNA-Binding Proteins in Plant Growth, Development, and Stress Responses. Mol. Cells 2016, 39, 179–185.
- Koster, T.; Marondedze, C.; Meyer, K.; Staiger, D. RNA-Binding Proteins Revisited—The Emerging Arabidopsis mRNA Interactome. Trends Plant Sci. 2017, 22, 512–526.
- Dedow, L.; Bailey-Serres, J. Searching for a Match: Structure, Function and Application of Sequence-Specific RNA-Binding Proteins. Plant Cell Physiol. 2019, 60, 1927–1938.
- Marondedze, C.; Thomas, L.; Serrano, N.; Lilley, K.; Gehring, C. The RNA-binding protein repertoire of Arabidopsis thaliana. Sci. Rep. 2016, 6, 29766.
- Castello, A.; Hentze, M.; Preiss, T. Metabolic Enzymes Enjoying New Partnerships as RNA-Binding Proteins. Trends Endocrinol. Metab. 2015, 26, 746–757.
- Hentz, M.; Preiss, T. The REM phase of gene regulation. Trends Biochem. Sci. 2010, 35, 423–426.
- Vermel, M.; Guermann, B.; Delage, L.; Grienenberger, J.; Maréchal-Drouard, L.; Gualberto, J. A family of RRM-type RNA-binding proteins specific to plant mitochondria. PNAS 2002, 99, 5866–5871.
- Staiger, D.; Zecca, L.; Wieczorek Kirk, D.; Apel, K.; Eckstein, L. The circadian clock regulated RNA-binding protein AtGRP7 autoregulates its expression by influencing alternative splicing of its own pre-mRNA. Plant J. 2003, 33, 361–371.
- Lee, J.; Lee, I. Regulation and function of SOC1, a flowering pathway integrator. J. Exp. Bot. 2010, 61, 2247–2254.
- Lin, C.; Miles, W. Beyond CLIP: Advances and opportunities to measure RBP–RNA and RNA–RNA interactions. Nucleic Acids Res. 2019, 47, 5490–5501.
- He, X.; Hsu, Y.; Zhu, S.; Wierzbicki, A.; Pontes, O.; Pikaard, C.; Liu, H.; Wang, C.; Jin, H.; Zhu, J. An effector of RNA-directed DNA methylation in Arabidopsis is an ARGONAUTE 4- and RNA-binding protein. Cell 2009, 137, 498–508.
- Gagliardi, M.; Matarazzo, M. RIP: RNA Immunoprecipitation. Methods Mol. Biol. 2016, 1480, 73–86.
- Zambelli, F.; Pavesi, G. RIP-Seq Data Analysis to Determine RNA-protein Associations. Methods Mol. Biol. 2015, 1269, 293–303.
- Niranjanakumari, S.; Lasda, E.; Brazas, R.; Garcia-Blanco, M. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 2002, 26, 182–190.
- Ule, J.; Jensen, K.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R. CLIP Identifies Nova-Regulated RNA Networks in the Brain. Science 2003, 302, 1212–1215.
- Licatalosi, D.; Mele, A.; Fak, J.; Ule, J.; Kayikci, M.; Chi, S.; Clark, T.; Schweitzer, A.; Blume, J.; Wang, X.; et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456, 464–469.
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.; Munschauer, M.; et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell 2010, 141, 129–141.
- König, J.; Zarnack, K.; Rot, G.; Curk, T.; Kayikci, M.; Zupan, B.; Turner, D.; Luscombe, N.; Ule, J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2014, 17, 909–915.
- Nostrand, E.; Pratt, G.; Shishkin, A.; Gelboin-Burkhart, C.; Fang, M.; Sundararaman, B.; Blue, S.; Nguyen, T.; Surka, C.; Elkins, K.; et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 2016, 13, 508–514.
- Kargapolova, Y.; Levin, M.; Lackner, K.; Danckwardt, S. sCLIP––An integrated platform to study RNA–protein interactomes in biomedical research: Identification of CSTF2tau in alternative processing of small nuclear RNAs. Nucleic Acids Res. 2017, 45, 6074–6086.
- Zarnegar, B.; Flynn, R.; Shen, Y.; Do, B.; Chang, H.; Khavari, P. irCLIP platform for efficient characterization of protein–RNA interactions. Nat. Methods 2015, 13, 489–492.
- Wang, T.; Xie, Y.; Xiao, G. dCLIP: A computational approach for comparative CLIP-seq analyses. Genome Biol. 2014, 15, R11.
- Ramanathan, M.; Porter, D.; Khavari, P. Methods to study RNA–protein interactions. Nat. Methods 2019, 16, 225–234.
- Darnell, R. HITS-CLIP: Panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA 2010, 1, 266–286.
- Maticzka, D.; Ilik, I.; Aktas, T.; Backofen, R.; Akhtar, A. uvCLAP is a fast and non-radioactive method to identify in vivo targets of RNA-binding proteins. Nat. Commun. 2018, 9, 1142.
- McMahon, A.; Rahman, R.; Jin, H.; Shen, J.; Fieldsend, A.; Luo, W.; Rosbash, M. TRIBE: Hijacking an RNA-Editing Enzyme to Identify Cell-Specific Targets of RNA-Binding Proteins. Cell 2016, 165, 742–753.
- Xu, W.; Rahman, R.; Rosbash, M. Mechanistic implications of enhanced editing by a HyperTRIBE RNA-binding protein. RNA 2018, 24, 173–182.
- Xing, D.; Wang, Y.; Hamilton, M.; Ben-Hur, A.; Reddy, A. Transcriptome-Wide Identification of RNA Targets of Arabidopsis SERINE/ARGININE-RICH45 Uncovers the Unexpected Roles of This RNA Binding Protein in RNA Processing. Plant Cell 2015, 27, 3294–3308.
- Zhang, X.; Shi, Y.; Powers, J.; Gowda, N.; Zhang, C.; Ibrahim, H.; Ball, H.; Chen, S.; Lu, H.; Mount, S. Transcriptome analyses reveal SR45 to be a neutral splicing regulator and a suppressor of innate immunity in Arabidopsis thaliana. BMC Genom. 2017, 18, 772.
- Albaqami, M.; Laluk, K.; Reddy, A. The Arabidopsis splicing regulator SR45 confers salt tolerance in a splice isoform-dependent manner. Plant Mol. Biol. 2019, 100, 379–390.
- Park, H.; You, Y.; Lee, A.; Jung, H.; Jo, S.; Oh, N.; Kim, H.; Lee, H.; Kim, J.; Kim, Y.; et al. OsFKBP20-1b interacts with the splicing factor OsSR45 and participates in the environmental stress response at the post-transcriptional level in rice. Plant J. 2020, 102, 992–1007.
- Francisco-Mangilet, A.; Karlsson, P.; Kim, M.; Eo, H.; Oh, S.; Kim, J.; Kulcheski, F.; Park, S.; Manavella, P. THO2, a core member of the THO/TREX complex, is required for microRNA production in Arabidopsis. Plant J. 2015, 82, 1018–1029.
- Streitner, C.; Danisman, S.; Wehrle, F.; Schoning, J.; Alfano, J.; Staiger, D. The small glycine-rich RNA binding protein AtGRP7 promotes floral transition in Arabidopsis thaliana. Plant J. 2008, 56, 239–250.
- Meyer, K.; Koster, T.; Nolte, C.; Weinholdt, C.; Lewinski, M.; Grosse, I.; Staiger, D. Adaptation of iCLIP to plants determines the binding landscape of the clockregulated RNA-binding protein AtGRP7. Genome Biol. 2017, 18, 204.
- Bardou, F.; Ariel, F.; Simpson, C.; Romero-Barrios, N.; Laporte, P.; Balzergue, S.; Brown, J.; Crespi, M. Long Noncoding RNA Modulates Alternative Splicing Regulators in Arabidopsis. Dev. Cell 2014, 30, 166–176.
- Bazin, J.; Romero, N.; Rigo, R.; Charon, C.; Blein, T.; Ariel, F.; Crespi, M. Nuclear Speckle RNA Binding Proteins Remodel Alternative Splicing and the Non-coding Arabidopsis Transcriptome to Regulate a Cross-Talk Between Auxin and Immune Responses. Front. Plant Sci. 2018, 9, 1209.
- Zhang, Y.; Gu, L.; Hou, Y.; Wang, L.; Deng, X.; Hang, R.; Chen, D.; Zhang, X.; Zhang, Y.; Liu, C.; et al. Integrative genome-wide analysis reveals HLP1, a novel RNA-binding protein, regulates plant flowering by targeting alternative polyadenylation. Cell Res. 2015, 25, 864–876.
- Yin, Q.; Cui, Y.; Zhang, G.; Zhang, H.; Wang, X.; Yang, Z. The Arabidopsis pentatricopeptide repeat protein PDM1 is associated with the intergenic sequence of S11-rpoA for rpoA monocistronic RNA cleavage. Chin. Sci. Bull. 2012, 57, 3452–3459.
- Tian, L.; Dorozhenk, K.; Zhang, L.; Fukuda, M.; Washida, H.; Kumamaru, T.; Okita, T. Zipcode RNA-Binding Proteins and Membrane Trafficking Proteins Cooperate to Transport Glutelin mRNAs in Rice Endosperm. Plant Cell 2020, 32, 2566–2581.
- Tian, L.; Chou, H.; Zhang, L.; Okita, T. Targeted Endoplasmic Reticulum Localization of Storage Protein mRNAs Requires the RNA-Binding Protein RBP-L. Plant Physiol. 2019, 179, 1111–1131.
- Juntawong, P.; Sorenson, R.; Bailey-Serres, J. Cold shock protein 1 chaperones mRNAs during translation in Arabidopsis thaliana. Plant J. 2013, 74, 1016–1028.
- Wierzbicki, A.; Ream, T.; Haag, J.; Pikaard, C. RNA Polymerase V transcription guides ARGONAUTE4 to chromatin. Nat. Genet. 2009, 41, 630–634.
- Zhu, Y.; Rowley, J.; Bohmdorfer, G.; Wierzbicki, A. A SWI/SNF Chromatin-Remodeling Complex Acts in Noncoding RNA-Mediated Transcriptional Silencing. Mol. Cell 2013, 49, 298–309.
- Rowley, J.; Bohmdorfer, G.; Wierzbicki, A. Analysis of long non-coding RNAs produced by a specialized RNA Polymerase in Arabidopsis thaliana. Methods 2013, 63, 160–169.
- Marmisolle, F.; García, M.; Reyes, C. RNA-binding protein immunoprecipitation as a tool to investigate plant miRNA processing interference by regulatory proteins of diverse origin. Plant Methods 2018, 14, 9.
- Schmid, L.; Ohler, L.; Mohlmann, T.; Brachmann, A.; Muino, J.; Leister, D.; Meurer, J.; Manavski, N. PUMPKIN, the Sole Plastid UMP Kinase, Associates with Group II Introns and Alters Their Metabolism. Plant Physiol. 2019, 179, 248–264.
- Libault, M.; Pingault, L.; Zogli, P.; Schiefelbein, J. Plant Systems Biology at the Single-Cell Level. Trends Plant Sci. 2017, 22, 949–960.
- Martin, L.; Nicolas, P.; Matas, A.; Shinozaki, Y.; Catalá, C.; Rose, J. Laser microdissection of tomato fruit cell and tissue types for transcriptome profiling. Nat. Protoc. 2016, 11, 2376–2388.
- Engreitz, J.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.; et al. The Xist lncRNA Exploits Three-Dimensional Genome Architecture to Spread across the X Chromosome. Science 2013, 341, 1237973.
- McHugh, C.; Chen, C.; Chow, A.; Surka, C.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236.
- Chu, C.; Qu, K.; Zhong, F.; Artandi, S.; Chang, H. Genomic Maps of Long Noncoding RNA Occupancy Reveal Principles of RNA-Chromatin Interactions. Mol. Cell 2011, 44, 667–678.
- Chu, C.; Zhang, Q.; Heard, E.; Chang, H. Systematic Discovery of Xist RNA Binding Proteins. Cell 2015, 161, 404–416.
- Foley, S.; Gosai, S.; Wang, D.; Selamoglu, N.; Sollitti, A.; Koster, T.; Steffen, A.; Lyons, E.; Daldal, F.; Garcia, B.; et al. A Global View of RNA-Protein Interactions Identifies Post-transcriptional Regulators of Root Hair Cell Fate. Dev. Cell 2017, 41, 204–220.
- Zhang, Z.; Sun, W.; Shi, T.; Lu, P.; Zhuang, M.; Liu, J. Capturing RNA–protein interaction via CRUIS. Nucleic Acids Res. 2020, 48, e52.
- Gemmill, D.; D’souza, S.; Meier-Stephenson, V.; Patel, T. Current approaches for RNA-labelling to identify RNA-binding proteins. Biochem. Cell. Biol. 2020, 98, 31–41.
- Johansson, H.; Liljas, L.; Uhlenbeck, O. RNA Recognition by the MS2 Phage Coat Protein. Semin. Virol. 1997, 8, 176–185.
- Lim, F.; Downey, T.; Peabody, D. Translational Repression and Specific RNA Binding by the Coat Protein of the Pseudomonas Phage PP7. J. Biol. Chem. 2001, 276, 22507–22513.
- Sun, H.; Tan, W.; Zu, Y. Aptamers: Versatile molecular recognition probes for cancer detection. Analyst 2016, 141, 403.
- Mallikaratchy, P. Evolution of Complex Target SELEX to Identify Aptamers against Mammalian Cell-Surface Antigens. Molecules 2017, 22, 215.
- Han, S.; Zhao, B.S.; Myers, S.; Carr, S.; He, C.; Ting, A. RNA–protein interaction mapping via MS2- or Cas13-based APEX targeting. PNAS 2020, 117, 22068–22079.
- Mukherjee, J.; Hermesh, O.; Eliscovich, C.; Nalpas, N.; Franz-Wachtel, M.; Macek, B.; Jansen, R.P. β-Actin mRNA interactome mapping by proximity biotinylation. PNAS 2019, 116, 12863–12872.
- Kaewsapsak, P.; Schechner, D.; Mallard, W.; Rinn, J.; Ting, A. Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. eLife 2017, 6, e29224.
- Yi, W.; Li, J.; Zhu, X.; Wang, X.; Fan, L.; Sun, W.; Liao, L.; Zhang, J.; Ye, J.; Chen, F.; et al. CRISPR-assisted detection of RNA–protein interactions in living cells. Nat. Methods 2020, 17, 685–688.
- Slavkovic, S.; Churcher, Z.; Johnson, P. Nanomolar binding affinity of quinine-based antimalarial compounds by the cocaine-binding aptamer. Bioorg. Med. Chem. 2018, 1, 5427–5434.
- Debiais, M.; Lelievre, A.; Smietana, M.; Muller, S. Splitting aptamers and nucleic acid enzymes for the development of advanced biosensors. Nucleic Acids Res. 2020, 48, 3400–3422.
- Bach-Pages, M.; Castello, A.; Preston, G. Plant RNA Interactome Capture: Revealing the Plant RBPome. Trends Plant Sci. 2017, 22, 449–451.
- Reichel, M.; Liao, Y.; Rettel, M.; Ragan, C.; Evers, M.; Alleaume, A.; Horos, R.; Hentze, M.; Preiss, T.; Millar, A. In Planta Determination of the mRNA-Binding Proteome of Arabidopsis Etiolated Seedlings. Plant Cell 2016, 28, 2435–2452.
- Zhang, Z.; Boonen, K.; Ferrari, P.; Schoofs, L.; Janssens, E.; van Noort, V.; Rolland, F.; Geuten, K. UV crosslinked mRNA-binding proteins captured from leaf mesophyll protoplasts. Plant Methods 2016, 12, 42.
- Marondedze, C.; Thomas, L.; Gehring, C.; Lilley, K. Changes in the Arabidopsis RNA-binding proteome reveal novel stress response mechanisms. BMC Plant Biol. 2019, 19, 139.
- Marondedze, C.; Thomas, L.; Lilley, K.; Gehring, C. Drought Stress Causes Specific Changes to the Spliceosome and Stress Granule Components. Front. Mol. Biosci. 2020, 6, 163.
- Perez-Perri, J.; Rogell, B.; Schwarzi, T.; Stein, F.; Zhou, Y.; Rettel, M.; Brosig, A.; Hentze, M. Discovery of RNA-binding proteins and characterization of their dynamic responses by enhanced RNA interactome capture. Nat. Commun. 2018, 9, 4408.
- Bach-Pages, M.; Homma, F.; Kourelis, J.; Kaschani, F.; Mohammed, S.; Kaiser, M.; van der Hoorn, R.; Castello, A.; Preston, G. Discovering the RNA-Binding Proteome of Plant Leaves with an Improved RNA Interactome Capture Method. Biomolecules 2020, 10, 661.
- Queiroz, R.; Smith, T.; Villanueva, E.; Marti-Solano, M.; Monit, M.; Pizzinga, M.; Mirea, D.; Ramakrishna, M.; Harvey, R.; Dezi, V.; et al. Comprehensive identification of RNA–protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat. Biotechnol. 2019, 37, 169–178.
- Liu, J.; Zhang, C.; Jia, X.; Wang, W.; Yin, H. Comparative analysis of RNA-binding proteomes under Arabidopsis thaliana-Pst DC3000-PAMP interaction by orthogonal organic phase separation. Int. J. Biol. Macromol. 2020, 160, 47–54.



