 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Simone Patergnani | + 2671 word(s) | 2671 | 2021-03-16 09:23:01 | | | |
| 2 | Camila Xu | Meta information modification | 2671 | 2021-03-18 03:27:30 | | |
Video Upload Options
Mitochondria are essential semi-autonomous cellular organelles with a double membrane composed by an inner (IMM) and an outer membrane (OMM).
1. Introduction
Free radicals and other reactive oxygen species (ROS) are frequently associated with being harmful. However, several physiological functions (differentiation, cellular signaling, phosphorylation/dephosphorylation events and apoptosis) are dependent on the presence of reactive species inside the cells [1]. To regulate these cellular functions, the ROS production must be kept low. When it increases, ROS become dangerous, determining undesirable effects on cellular structures, including intracellular organelles, and participating in the onset and/or progression of several human disorders, such as neurodegenerative disorders, cancer, pulmonary, and cardiovascular diseases [2][3][4][5]. ROS can be produced at cytosolic levels by the NADPH oxidase (NOX) and the nitric oxide (NO) synthase enzyme. The well-characterized source of cytosolic ROS is NOX2, which produces anion superoxide (O2·−) through NAPDH electron exchange [6]. Alternatively, significant amounts of ROS are generated in the peroxisomes compartment by oxidases and NO synthases that produce hydrogen peroxide (H2O2) and NO, respectively [7]. The endoplasmic reticulum (ER) represents a place where high amounts of ROS are produced. In this compartment, ROS result in being the byproduct of oxidases and oxygenase involved during the protein folding process, such as ER oxidoreductin 1 [8]. Moreover, ER present iron deposits that contribute to form the reactive species hydroxyl radical (·OH), but undoubtedly, mitochondria represent the primary source of ROS (mitochondrial ROS, mtROS) for the cell [9].
Mitochondria are essential semi-autonomous cellular organelles with a double membrane composed by an inner (IMM) and an outer membrane (OMM). OMM separates the mitochondria from cytoplasm and it may be freely traversed by many proteins (5000 daltons or less), ions, such as Calcium (Ca2+), and metabolites, while the larger molecules are imported by specific translocase [10][11][12]. IMM is highly specialized and permits the passage of small molecules, including oxygen, carbon dioxide, and water. The passage throughout IMM of any other macromolecules is permitted by the action of dedicated translocase, like TIM (translocase of the inner membrane). Within the organelle, OMM and IMM allow the formation of two different compartments: the intermembrane space (IMS) and the matrix [13]. IMS plays a crucial role in regulating different cellular activities, such as mitochondrial respiration, proteins transport, lipid homeostasis and metal ion exchange; while the matrix contains the mitochondrial DNA (mtDNA), mitochondrial ribosomes and multiple diverse metabolic pathways [tricarboxylic acid (TCA) cycle, β-oxidation, and heme synthesis], that modulate the assembly and genes expression, as well as, Ca2+ uptake which is critically important to cellular function [14][15][16] mtDNA is a closed circle double-stranded DNA, without histones and of effective repair mechanism. mtDNA encodes for 37 genes, including 13 components of the mitochondrial electron transport chain (mETC) [17]. Mitochondrial matrix is also the site of a series of enzyme that convert pyruvate and fatty acid in acetyl-CoA. Once produced, acetyl-CoA enters in TCA cycle which produces NADH and FADH2 that are used by mETC [18].
mETC is composed by four complexes (I-IV) that are embedded in the IMM and are responsible to create the electrochemical gradient required to generate ATP. Notably, complex-I (NADH-CoQ reductase) and complex III (cytochrome c reductase) represent the primary sites of production of mitochondrial O2·− (Figure 1). This byproduct of the mitochondrial respiration is also considered the most quantitatively important mtROS source in higher organisms [2]. However, mitochondria are not only an ROS-producing compartment but, in turn, are their targets. In particular, mitochondrial phospholipids and DNA are susceptible to mtROS-induced oxidative damage (Figure 2). Mitochondrial phospholipids loss their permeability and fluidity compromising the functioning of all factor associated to the membrane, in particular mETC members and ion channels. mtDNA loses its integrity, acquiring mutations and reducing in the number of mtDNA copies. In turn, mtDNA mutations can determine further alteration in mitochondrial functioning and signaling that may be determinant for various human diseases [19]. To avoid accumulating oxidative damages, mitochondria have evolved different mechanisms to reduce the mtROS-induced oxidative damage or eliminate injured mitochondria. The mitochondrial antioxidant systems and the mitochondrial stress responses (including mitophagy, mitochondrial unfolded protein response (mtUPR), and apoptosis) represent some classical examples [2]. In addition, it is widely accepted that alterations in mitochondrial functions (like in Ca2+ dynamics and/or in lipid transfer from ER to mitochondria) modulate the mtROS production [2]. Thus, increased levels of mitochondrial Ca2+ activate ROS-generating enzyme and the formation of radicals.
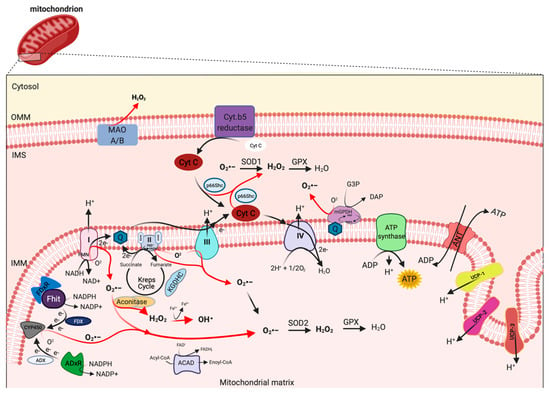
Figure 1. Mitochondrial sites of ROS production. Mitochondrial complex I and III of respiratory chain are the principal sites of O2•− production within a cell, which can be converted to H2O2 by superoxide dismutase (SOD1 and SOD2) enzymes. H2O2 in turn is rapid neutralized to H2O and oxygen by glutathione peroxidase (GPX). However, other mitochondrial proteins, localized from OMM to matrix, may also contribute to mtROS production, including monoamine oxidase A and B (MAO A/B), cytochrome (Cyt.) b5 reductase, mitochondrial glycerol-phosphate dehydrogenase (mGPDH), p66Shc, Fhit with ferredoxin reductase (FDxR), adrenodoxin reductase (ADxR)-adrenodoxin (ADX)-cytochrome P450scc (CYP450) system, α-ketoglutharate dehydrogenases (KGDHC), acyl-CoA dehydrogenases (ACAD) and aconitase. This figure has been created with “BioRender.com.”.
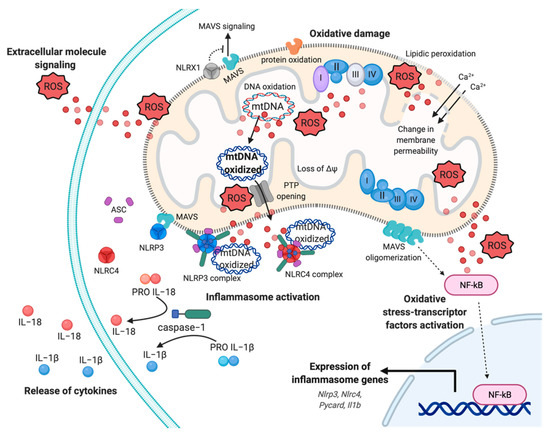
Figure 2. Mitochondrial ROS-induced inflammatory response. Representation of multifaceted aspects of mitochondrial ROS in inflammation. Increased mtROS cause oxidative damage to mitochondrial membrane, with events of lipidic peroxidation and changes in membrane permeability, molecules, proteins, and mtDNA, which contribute to mitochondrial dysfunction and exacerbation of mtROS production. The dissemination of mtROS actives the redox-sensitive transcription factor NF-kB, inducing the expression of inflammasome genes, such as Nlrp3, Nlrc4, and Il1b genes. In turn, the mtROS and mtDNA promote the cytokines release mediating the inflammasome NLRP3 and NLRC4 complex activation, through the recruitment of pro-caspase−1 and the processing of pro-IL−1 and pro-IL−18. Finally, mtROS are reversed to extracellular milieu to sustain and exacerbate the inflammatory responses, affecting proximal cells and conditioning the inflammatory microenvironment. Abbreviations: Reactive oxygen species, ROS; Interleukin−18, IL−18; Interleukin−1, IL−1; NLR Family CARD Domain Containing 4, NLRC4; ASC; NLR Family Pyrin Domain Containing 3, NLRP3; Mitochondrial antiviral-signaling protein, MAVS; mitochondrial deoxyribonucleic acid, mtDNA; NLR Family member X1 precursor, NLRX1; Calcium, Ca2+; Nuclear factor kappa-light-chain-enhancer of activated B cells, NF-kB; Mitochondrial membrane potential, ΔΨ. This figure has been created with “BioRender.com.”.
Clearly, mitochondria and mtROS production are primary signal hub for the cells. It is not surprising that diverse studies highlight their involvement in pathogenesis of various diseases, including neurodegenerative disorders, cancer, viral and bacterial infection, cardiovascular diseases, metabolic syndromes, and autoimmune disorders [20]. In particular, it has been proposed that mitochondrial dysfunction and excessive mtROS levels sustain inflammation in these pathological conditions. In these contexts, mtROS-induced inflammation acts as a feedback system creating a stressful environment, where the exacerbation of inflammation provokes tissue damage and becomes a chronic event [20]. Considering the importance of oxidative stress in inflammation, it is easy to speculate that therapeutic manipulations aimed to prevent oxidative damage, targeting the mitochondrial (dys)function and generating an antioxidant power, may represent an opportunity to disrupt the reciprocal relation between mtROS and “mito-inflammation”. A new concept that identifies the compartmentalization of the inflammatory process in which the mitochondrion acts as central regulator, checkpoint, and arbitrator.
2. Mitochondria: A ROS Production Machinery
Mitochondria are widely recognized as a source of ROS production within most mammalian cells in both physiological and pathological conditions. The generation of mtROS from mitochondria was first discovered during the early 1970s [21][22]. According to estimates, 1–2% of total cellular oxygen consumption is going to ROS production [23].
mtROS are produced as byproduct of bioenergetic metabolism during the process of oxidative phosphorylation (OXPHOS) and formed by one-electron transfers, generating O2•− that can be converted to H2O2 by superoxide dismutase (SOD) enzymes [24]. Multiple sites of mtROS production have long been identified in the organelle and mtROS can take place both in the mitochondrial matrix by the core metabolic machinery present in the IMM and in the intermembrane space [25]. Generation of mtROS mainly takes place at the ETC, located on the IMM, during the process of OXPHOS [26], which includes the major sites of the respiratory chain Complex I (NADH dehydrogenase (ubiquinone), 45 protein subunits), Complex III (ubiquinol-cytochrome c reductase, 10 proteins subunits), and also the dihydrolipoamide dehydrogenase enzyme [27][28]. Complex I generates O2•− by reducing flavin mononucleotide (FMN) site on complex I and reversing electron transfer from the coenzyme Q (CoQ) pool back to complex I [2]. Basically, complex II (succinate dehydrogenase) was not considered a source of ROS per se, instead its contribution to ROS generation is linked to reverse electron transfer, in which electrons are transferred from succinate to ubiquinone through complex II and then back to complex I [29]. Complex II consists of four subunits. Two subunits are located on the matrix side of the IMM: SDHA (succinate dehydrogenase), the flavoprotein subunit covalently bound a FAD cofactor, which removes electrons from succinate; SDHB, the iron-sulfur protein subunit, contains the binding site of the substrate succinate, three clusters (2Fe-2S), (4Fe-4S), and (3Fe-4S) that mediate electron transfer to the ubiquinone molecule; and SDHC and SDHD the two anchor subunits to the IMM, the assembly factors participate in the biogenesis of complex II [30][31][32]. Brand’s group demonstrated that complex II can produce superoxide through flavin adenine nucleotide (FAD) [33]. Indeed, it has been suggested that other less well-described sites may also participates in ROS production including, the electron transferring flavoprotein/ETF:Q oxidoreductase (ETF/ETF:QOR) system of fatty acid β-oxidation [34], dihydroorotate dehydrogenase [35], and proline dehydrogenase [36]. These different mtROS’s sites have distinct signaling roles and subsequently the primary production sites change under different physiological conditions [37]. Indeed, the production of radicals from the mitochondrial respiratory chain is conditioned by multiple factors, including mitochondrial membrane potential, metabolic state of mitochondria and oxygen levels [38]. Other mitochondrial proteins may participate to increment the mtROS production. In particular, the mitochondrial enzyme dehydrogenase of α phosphate dehydrogenase (glycerol α phosphate dehydrogenase, mGPDH), located at the outer surface of the IMM, is a flavoprotein containing FAD and serves as an electron shuttle linking the cytosolic NADH/ NAD recycling to the mETC implicated in lipid metabolism capable of producing cytosolic NAD+ from the NADH formed in glycolysis [39]. However, it is unequally expressed in numerous mouse tissues mediating the generation of H2O2 [40]. Therefore, other potential sources of mtROS are still more poorly explored, such as acyl-CoA dehydrogenases (ACAD), which are flavoproteins and have increasingly been recognized as oxidant sources in mitochondria, involved in lipid catabolism [41]. Both of these proteins are implicated in mtROS production in tissues during the oxidation lipid-derived substrates [42]. The mitochondrial enzyme adrenodoxin reductase (ADxR)-adrenodoxin (ADX)-cytochrome P450scc (CYP450) system (cholesterol side chain cleavage) is also involved in superoxide generation and is coupled with NADPH in the mitochondrial matrix [43]. The onco-suppressor Fhit protein, imported into mitochondria, interacts with ferredoxin reductase (FDxR), which transfers electrons from NADPH to CYP450 via ferredoxin (FDX), increasing the intracellular superoxide production [44][45]. The redox enzyme, p66shc, regulates the oxidative stress acting a different level. This adaptor protein may induce ROS generation by: (i) Translocating into mitochondria, after PKC-dependent phosphorylation, to transfer electrons from reduced cytochrome C to oxygen [46][47], (ii) activating the Rac-1-dependent plasma membrane NADPH oxidase [48], and (iii) downregulating the expression of antioxidant enzymes, such as glutathione peroxidase-1 and manganese superoxide dismutase (Mn-SOD) [49][50]. The enzymes monoamine oxidase (MAO-A and MAO-B) are located in the OMM and expressed in various mammalian tissues catalyze the oxidation of biogenic amines accompanied by the release of H2O2 [51]. Mitochondrial aconitase is an enzyme positioned in the matrix, the enzyme contains an iron-sulfur cluster that can be oxidized by O2•− or H2O2 generating •OH [52].
Uncoupling proteins (UCPs) are IMM proteins that belong to a family of mitochondrial transporters regulating the proton transport across the IMM. In particular, UCPs determine an inducible proton leak by causing mild-uncoupling events. During mild-uncoupling process, the mitochondrial membrane potential (ΔΨ) and the reduction events in the ETC diminished, thereby resulting in decreases in ROS production from the ETC [53].
UCPs exists in three forms: UCP 1-3. UCP1 is primary found in mitochondria from brown adipose tissue, where it regulates the thermogenesis in response to cold exposure and modulates the ROS production. Consistently, UCP1 knockdown mice are characterized by low levels of ΔΨ, reduced O2 consumption rate, and increased ROS production [54]. In addition, it has been recently demonstrated that UCP1 regulates the ROS production also in kidney. Deletion of UCP1 results in increased oxidative stress in kidney and exacerbates an ischemic condition or a nephrotoxic drug-induced acute kidney injury. Contrarily, viral-based expression of UCP1 suppresses the mtROS production and alleviates the induced kidney injury [55]. UCP2 and UCP3 mRNA expression are ubiquitous and their proteins are present in different tissues, in particular skeletal muscle, central nervous system, pancreas, and spleen. UCP2 and UCP3 protect mitochondrial proteins from endogenous ROS (such as O2•−), and UCP3 and UCP2 knockout models present increased ROS levels. In addition, it has been demonstrated that ROS, 4-hydroxy-2-nonenal (4-HNE), and lipid peroxidation determine activation of UCPs [56][57]. These findings suggest that UCPs exist in a feed-back mechanism, where increased ROS production activates a protective mechanism (uncoupling events) necessary to reduce the ROS formation and the consequent ROS damage. UCP2 has also an important role during insulin secretion in pancreatic β cells. In fact, UCP2 contributes to regulate the intracellular ROS levels of β cells, thereby controlling the excessive glucose-stimulated insulin secretion (GSIS) [58]. UCP2 represent an essential metabolite transporter that regulates the mitochondrial export into the cytosol of metabolites: malate, oxaloacetate and aspartate. Because of this transport, UCP2 prevents the oxidation of glucose and sustains the glutaminolysis [59]. Another protein that preserves an efficient electron transport and control the ROS production is hexokinase (HK) 2. HK2 is recruited to the OMM where it binds itself to the ADP/ATP exchange complex formed by VDAC and ANT. The role of HK2 is to provide continuously ADP in order to generate the ADP/ATP recycling mechanism essential to preserve the optimal respiration rate, thereby preventing the dangerous electron leak producing ROS [60]. In line with this evidence, HK2 knockdown and the dissociation of HK2 from mitochondria increase ROS production and activate the mitochondrial permeability transition (MPT) both in vivo and in vitro [61]. Accordingly, HK2 overexpression decreases the mtROS levels [61].
Sirtuins (SIRTs) are nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylases that play pivotal role in diverse cellular functions, including protein acetylation and deacetylation, metabolism, mitochondrial functioning, and cell survival. In addition, all seven SIRTs isoforms have been associated to process related to antioxidant and redox signaling. In particular, the functions of SIRT1 (the most well-studied member) have been associated to mediate protection from ROS. At demonstration, small molecule activators of SIRT1, such as SRT1720, determine activation of SOD2 levels, accompanied by reduction in the formation of 4-HNE and in protein carbonylation levels [62]. SIRT1 can be also directly activated by oxidative stress and is required for DNA repairing mechanisms following H2O2-induced damage [63]. Similarly, the antioxidant compound resveratrol activates SIRT1 to protect cells from H2O2-induced cell death [64]. Another important SIRTs modulating ROS levels is SIRT3. Downregulation of SIRT3 is associated with increases of ROS production and activation of the protumorigenic transcription factor HIF-1α [65]. Furthermore, SIRT3 may activate the human 8-oxoguanine-DNA glycosylase 1 (OGG1), which is an enzyme crucial for the repairing of the mtDNA [66].
However, the levels of mtROS are conditioned by the specific action of mitochondrial antioxidant systems, recruited to detox the radicals produced. SODs are the antioxidant enzymes that convert O2•− to H2O2 (Figure 1). Two isoforms control the level of O2•−: SOD1/copper-zinc superoxide dismutase (Cu, Zn-SOD) and SOD2/ Mn-SOD isoform. SOD1 is widely distributed throughout the cell, cytoplasm, nucleus, and intermembrane space of mitochondria (IMS), whereas SOD2 is expressed only in the mitochondrial matrix [67] (Figure 1). The rapid conversion into H2O2 is counteract by catalase and glutathione peroxidase (GPX), which neutralize H2O2 in H2O and oxygen [68][69] (Figure 1). Catalase is mainly located to cytosol, indicating that the scavenge capability in mitochondria it is leaved to GPX [70]. So far, in mammals, there have been eight GPX isoforms identified, where GPX1-4 and GPX6 are selenoproteins with a selenocysteine as catalytic moiety and only GPX1 and GPX4 are expressed to mitochondria. The GPX-dependent scavenge capability is associated to use glutathione (GSH) as cofactor and electron source to neutralize H2O2.
References
- Kaludercic, N.; Deshwal, S.; Di Lisa, F. Reactive oxygen species and redox compartmentalization. Front. Physiol. 2014, 5, 285.
- Rimessi, A.; Previati, M.; Nigro, F.; Wieckowski, M.R.; Pinton, P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 2016, 81 Pt B, 281–293.
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262.
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv. Med. 2016, 2016, 9152732.
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197.
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902.
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta 2006, 1763, 1755–1766.
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Ero1alpha regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid. Redox Signal. 2012, 16, 1077–1087.
- Fan, Y.; Simmen, T. Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism. Cells 2019, 8, 1071.
- Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. J. Clin. Med. 2020, 9, 740.
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323.
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730.
- Kuhlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89.
- Hernandez-Aguilera, A.; Rull, A.; Rodriguez-Gallego, E.; Riera-Borrull, M.; Luciano-Mateo, F.; Camps, J.; Menendez, J.A.; Joven, J. Mitochondrial dysfunction: A basic mechanism in inflammation-related non-communicable diseases and therapeutic opportunities. Mediat. Inflamm. 2013, 2013, 135698.
- Du Toit, A. Transcription: A mitochondrial switch between transcription and replication. Nat. Rev. Mol. Cell Biol. 2015, 16, 128.
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723.
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol. 2019, 17, 53.
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta 2014, 1837, 418–426.
- Hahn, A.; Zuryn, S. Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants 2019, 8, 392.
- Sandhir, R.; Halder, A.; Sunkaria, A. Mitochondria as a centrally positioned hub in the innate immune response. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1090–1097.
- Loschen, G.; Flohe, L.; Chance, B. Respiratory chain linked H(2)O(2) production in pigeon heart mitochondria. FEBS Lett 1971, 18, 261–264.
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973, 134, 707–716.
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230.
- Nohl, H.; Gille, L.; Staniek, K. Intracellular generation of reactive oxygen species by mitochondria. Biochem. Pharmacol. 2005, 69, 719–723.
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214.
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19.
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414.
- Turrens, J.F.; Boveris, A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 1980, 191, 421–427.
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787.
- Sun, F.; Huo, X.; Zhai, Y.; Wang, A.; Xu, J.; Su, D.; Bartlam, M.; Rao, Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 2005, 121, 1043–1057.
- Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Pacak, K.; Neuzil, J. Mitochondrial Complex II: At the Crossroads. Trends Biochem. Sci. 2017, 42, 312–325.
- Hao, H.X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.P.; Kunst, H.; Devilee, P.; Cremers, C.W.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009, 325, 1139–1142.
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264.
- Seifert, E.L.; Estey, C.; Xuan, J.Y.; Harper, M.E. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J. Biol. Chem. 2010, 285, 5748–5758.
- Orr, A.L.; Quinlan, C.L.; Perevoshchikova, I.V.; Brand, M.D. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J. Biol. Chem. 2012, 287, 42921–42935.
- White, T.A.; Krishnan, N.; Becker, D.F.; Tanner, J.J. Structure and kinetics of monofunctional proline dehydrogenase from Thermus thermophilus. J. Biol. Chem. 2007, 282, 14316–14327.
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312.
- Ballinger, S.W. Mitochondrial dysfunction in cardiovascular disease. Free Radic. Biol. Med. 2005, 38, 1278–1295.
- Mracek, T.; Drahota, Z.; Houstek, J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim. Biophys. Acta 2013, 1827, 401–410.
- Jesina, P.; Kholova, D.; Bolehovska, R.; Cervinkova, Z.; Drahota, Z.; Houstek, J. Glycerophosphate-dependent hydrogen peroxide production by rat liver mitochondria. Physiol. Res. 2004, 53, 305–310.
- Cardoso, A.R.; Chausse, B.; da Cunha, F.M.; Luevano-Martinez, L.A.; Marazzi, T.B.; Pessoa, P.S.; Queliconi, B.B.; Kowaltowski, A.J. Mitochondrial compartmentalization of redox processes. Free Radic. Biol. Med. 2012, 52, 2201–2208.
- Lambertucci, R.H.; Hirabara, S.M.; Silveira Ldos, R.; Levada-Pires, A.C.; Curi, R.; Pithon-Curi, T.C. Palmitate increases superoxide production through mitochondrial electron transport chain and NADPH oxidase activity in skeletal muscle cells. J. Cell. Physiol. 2008, 216, 796–804.
- Hanukoglu, I.; Rapoport, R.; Weiner, L.; Sklan, D. Electron leakage from the mitochondrial NADPH-adrenodoxin reductase-adrenodoxin-P450scc (cholesterol side chain cleavage) system. Arch. Biochem. Biophys. 1993, 305, 489–498.
- Rimessi, A.; Marchi, S.; Fotino, C.; Romagnoli, A.; Huebner, K.; Croce, C.M.; Pinton, P.; Rizzuto, R. Intramitochondrial calcium regulation by the FHIT gene product sensitizes to apoptosis. Proc. Natl. Acad. Sci. USA 2009, 106, 12753–12758.
- Trapasso, F.; Pichiorri, F.; Gaspari, M.; Palumbo, T.; Aqeilan, R.I.; Gaudio, E.; Okumura, H.; Iuliano, R.; Di Leva, G.; Fabbri, M.; et al. Fhit interaction with ferredoxin reductase triggers generation of reactive oxygen species and apoptosis of cancer cells. J. Biol. Chem. 2008, 283, 13736–13744.
- Pinton, P.; Rimessi, A.; Marchi, S.; Orsini, F.; Migliaccio, E.; Giorgio, M.; Contursi, C.; Minucci, S.; Mantovani, F.; Wieckowski, M.R.; et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 2007, 315, 659–663.
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233.
- Khanday, F.A.; Santhanam, L.; Kasuno, K.; Yamamori, T.; Naqvi, A.; Dericco, J.; Bugayenko, A.; Mattagajasingh, I.; Disanza, A.; Scita, G.; et al. Sos-mediated activation of rac1 by p66shc. J. Cell Biol. 2006, 172, 817–822.
- Wu, Z.; Rogers, B.; Kachi, S.; Hackett, S.F.; Sick, A.; Campochiaro, P.A. Reduction of p66Shc suppresses oxidative damage in retinal pigmented epithelial cells and retina. J. Cell. Physiol. 2006, 209, 996–1005.
- Haga, S.; Terui, K.; Fukai, M.; Oikawa, Y.; Irani, K.; Furukawa, H.; Todo, S.; Ozaki, M. Preventing hypoxia/reoxygenation damage to hepatocytes by p66(shc) ablation: Up-regulation of anti-oxidant and anti-apoptotic proteins. J. Hepatol. 2008, 48, 422–432.
- Kunduzova, O.R.; Bianchi, P.; Parini, A.; Cambon, C. Hydrogen peroxide production by monoamine oxidase during ischemia/reperfusion. Eur. J. Pharmacol. 2002, 448, 225–230.
- Vasquez-Vivar, J.; Kalyanaraman, B.; Kennedy, M.C. Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J. Biol. Chem. 2000, 275, 14064–14069.
- Brand, M.D.; Affourtit, C.; Esteves, T.C.; Green, K.; Lambert, A.J.; Miwa, S.; Pakay, J.L.; Parker, N. Mitochondrial superoxide: Production, biological effects, and activation of uncoupling proteins. Free Radic. Biol. Med. 2004, 37, 755–767.
- Dlaskova, A.; Clarke, K.J.; Porter, R.K. The role of UCP 1 in production of reactive oxygen species by mitochondria isolated from brown adipose tissue. Biochim. Biophys. Acta 2010, 1797, 1470–1476.
- Jia, P.; Wu, X.; Pan, T.; Xu, S.; Hu, J.; Ding, X. Uncoupling protein 1 inhibits mitochondrial reactive oxygen species generation and alleviates acute kidney injury. EBioMedicine 2019, 49, 331–340.
- Echtay, K.S.; Esteves, T.C.; Pakay, J.L.; Jekabsons, M.B.; Lambert, A.J.; Portero-Otin, M.; Pamplona, R.; Vidal-Puig, A.J.; Wang, S.; Roebuck, S.J.; et al. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003, 22, 4103–4110.
- Murphy, M.P.; Echtay, K.S.; Blaikie, F.H.; Asin-Cayuela, J.; Cocheme, H.M.; Green, K.; Buckingham, J.A.; Taylor, E.R.; Hurrell, F.; Hughes, G.; et al. Superoxide activates uncoupling proteins by generating carbon-centered radicals and initiating lipid peroxidation: Studies using a mitochondria-targeted spin trap derived from alpha-phenyl-N-tert-butylnitrone. J. Biol. Chem. 2003, 278, 48534–48545.
- Robson-Doucette, C.A.; Sultan, S.; Allister, E.M.; Wikstrom, J.D.; Koshkin, V.; Bhattacharjee, A.; Prentice, K.J.; Sereda, S.B.; Shirihai, O.S.; Wheeler, M.B. Beta-cell uncoupling protein 2 regulates reactive oxygen species production, which influences both insulin and glucagon secretion. Diabetes 2011, 60, 2710–2719.
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965.
- Da-Silva, W.S.; Gomez-Puyou, A.; de Gomez-Puyou, M.T.; Moreno-Sanchez, R.; De Felice, F.G.; de Meis, L.; Oliveira, M.F.; Galina, A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 2004, 279, 39846–39855.
- Wu, R.; Wyatt, E.; Chawla, K.; Tran, M.; Ghanefar, M.; Laakso, M.; Epting, C.L.; Ardehali, H. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol. Med. 2012, 4, 633–646.
- Mitchell, S.J.; Martin-Montalvo, A.; Mercken, E.M.; Palacios, H.H.; Ward, T.M.; Abulwerdi, G.; Minor, R.K.; Vlasuk, G.P.; Ellis, J.L.; Sinclair, D.A.; et al. The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep. 2014, 6, 836–843.
- Abdelmohsen, K.; Pullmann, R., Jr.; Lal, A.; Kim, H.H.; Galban, S.; Yang, X.; Blethrow, J.D.; Walker, M.; Shubert, J.; Gillespie, D.A.; et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol. Cell 2007, 25, 543–557.
- Ido, Y.; Duranton, A.; Lan, F.; Weikel, K.A.; Breton, L.; Ruderman, N.B. Resveratrol prevents oxidative stress-induced senescence and proliferative dysfunction by activating the AMPK-FOXO3 cascade in cultured primary human keratinocytes. PLoS ONE 2015, 10, e0115341.
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 2011, 19, 416–428.
- Cheng, Y.; Ren, X.; Gowda, A.S.; Shan, Y.; Zhang, L.; Yuan, Y.S.; Patel, R.; Wu, H.; Huber-Keener, K.; Yang, J.W.; et al. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013, 4, e731.
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393.
- Kirkman, H.N.; Gaetani, G.F. Mammalian catalase: A venerable enzyme with new mysteries. Trends Biochem. Sci. 2007, 32, 44–50.
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303.
- Panday, S.; Talreja, R.; Kavdia, M. The role of glutathione and glutathione peroxidase in regulating cellular level of reactive oxygen and nitrogen species. Microvasc. Res. 2020, 131, 104010.

