 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Norbert Nemeth | + 5995 word(s) | 5995 | 2021-03-01 07:48:33 | | | |
| 2 | Rita Xu | -1822 word(s) | 4173 | 2021-03-17 04:35:08 | | |
Video Upload Options
Hepatic ischemia-reperfusion injury (IRI) is a multifactorial phenomenon which has been associated with adverse clinical outcomes. IRI related tissue damage is characterized by various chronological events depending on the experimental model or clinical setting. Despite the fact that IRI research is in the spotlight of scientific interest for over three decades with a significant and continuous increase in publication activity over the years and the large number of pharmacological and surgical therapeutic attempts introduced, not many of these strategies have made their way to the everyday clinical practice. Furthermore, the pathomechanism of hepatic IRI has not been fully elucidated yet. In the complex process of the IRI flow properties of blood are not neglectable. Hemorheological factors play an important role in determining tissue perfusion and orchestrating mechanical shear stress-dependent endothelial functions. Antioxidant and anti-inflammatory agents, ischemic conditioning protocols, dynamic organ preservation techniques may improve rheological properties of the post-reperfusion hepatic blood flow and target endothelial cells, exerting a potent protection against hepatic IRI.
1. Introduction
Liver ischemia-reperfusion injury (IRI) remains inevitable during extended liver resections and orthotopic liver transplantation (OLT). Hepatic IRI is a multifactorial phenomenon which has been associated with adverse clinical outcomes [1][2][3][4].
Ischemia-reperfusion injury-related tissue damage is characterized by various chronological events depending on the experimental model or clinical setting [4][5][6]. In major hepatectomies, a shorter phase of warm ischemia (e.g., intermittent or continuous Pringle/Baron maneuver) is followed by normothermic in situ reperfusion. In the setting of OLT, the extent of IR-related damage is significantly dependent on the type of organ donation. In a donation after brain death (DBD) cold ischemia is followed by a warm reperfusion in the recipient. The quality of the allograft, the length of cold storage and the time required for the implantation of the allograft are major determinants of clinical outcomes, especially in marginal DBD allografts [4]. In donation after circulatory death (DCD) donors, an additional period of in situ warm ischemia in the donor exposes the recipients to a higher risk of graft-related complications such as an increased risk of biliary complications (biliary stenosis, ischemic-type biliary lesions) [4].
The extent of IRI has been associated with a broad spectrum of complications and inferior outcomes in OLT and partial hepatectomies. IRI has been demonstrated to be a major risk-factor for perioperative morbidity, early allograft dysfunction (EAD) and primary non-function in liver transplantation [4]. Besides its short-term effects, IRI also has long-term consequences leading to graft injury, fibrosis, rejection and biliary injury [3][7] (Figure 1).
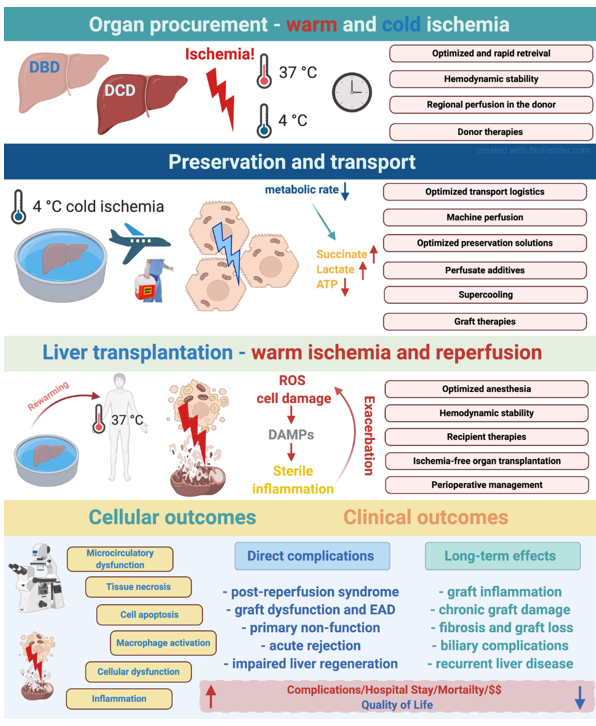
Figure 1. Various phases of ischemia reperfusion injury and their effects and mechanisms in the setting of deceased donor liver transplantation. Potential stages and interventional strategies to mitigate IRI during organ transplantation logistics. The figure was created with BioRender (https://biorender.com).
IRI research has continuously drawn scientific interest with a significant and continuous increase in publication activity over the years (Figure 2) and a large number of pharmacological and surgical therapeutic attempts suggested [4][5][8][9][10]. Nonetheless, few therapeutic interventions have made their way into everyday clinical practice.
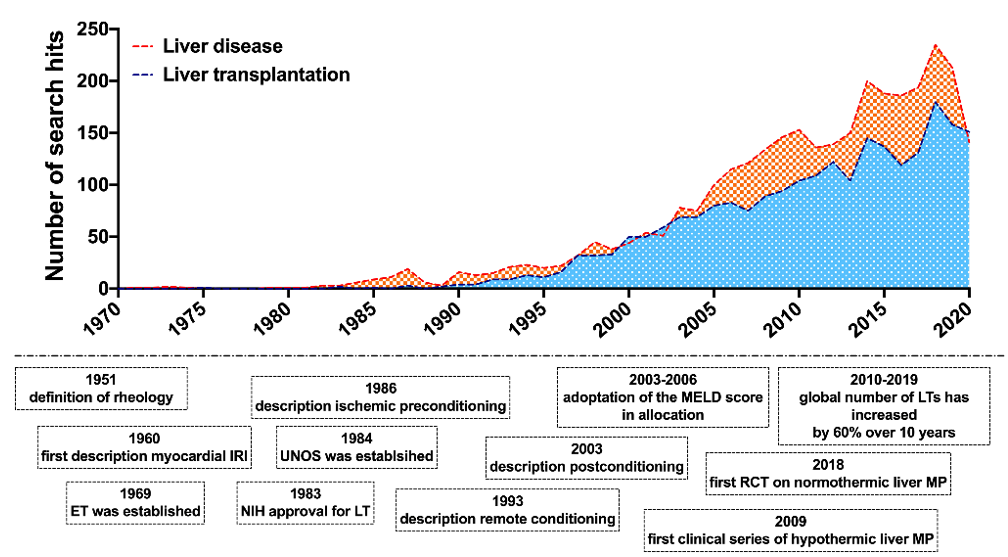
Figure 2. Major milestones in the history of hemorheology, ischemia-reperfusion and liver transplantation research. Publication activity in the field of rheological and ischemia-reperfusion injury research in liver disease and liver transplantation. Number of corresponding search hits in liver disease and liver transplantation in the PubMed® database. Accessed on 13th December 2020; search syntax: ((ischemia-reperfusion) OR (rheology)) AND (liver transplantation) // ((ischemia-reperfusion) OR (rheology)) AND (liver disease).
In recent decades extensive research has been conducted on cellular lesions and various signaling processes, from inflammatory mechanisms, and immune responses to metabolic and systemic changes, including alterations in remote organs’ function [1][11][12][13][14][15][16][17][18][19][20][21]. There is also growing knowledge about hemodynamic changes and the dynamics of microcirculation in general, however, there is another factor about which is discussed relatively little, and that is the rheology of the circulating blood itself [22][23][24][25][26][27]. Through its macro- and micro-rheological properties, blood plays an important role in modulating not only the determinant factors of tissue perfusion but also the shear stress mediated processes in the endothelial cells [28][29][30][31]. Accordingly, these are the elements of the complex etiologic mechanism of ischemia-reperfusion events that cannot be ignored.
2. Rheology of the Blood—A Brief Overview
Circulation, tissue perfusion, endothelial/vascular function: all are strongly linked to the rheological features of the blood [32]. The main parameters determining blood viscosity are the plasma viscosity, the hematocrit, and the micro-rheological factors, such as red blood cell deformability and aggregation [23][24][25]. All of these affect flow conditions and perfusion, showing a great complexity in the circulatory bed. Pressure (pressure gradient), shear rate—shear stress, velocity of flow, vascular resistance and impedance count in biomechanics of the blood stream. Several factors influence the blood flow in vivo, and estimating the flow conditions in large vessels using the major hydrodynamic and physical fluid flow equations can be only partially used. Therefore, a single constitutive equation is not appropriate to fully understand blood rheology. However, several approaches exist to define these conditions (e.g., Newtonian and non-Newtonian fluid, the Einstein model, Bingham fluid model, Casson model, Quemada model) [24][25][33].
2.1. Biomechanical, Cellular and Molecular Aspects
In non-Newtonian fluids, such as the blood, the viscosity increases with decreasing shear rate (the relation of shear stress and shear rate is non-linear: Casson-curve). Furthermore, in blood there is a minimal shear stress that is required to start the flow. It is called yield-stress. It is well known that the shear rate and shear stress significantly vary along the circulation with the highest shear stress occurring in the arterioles and capillaries [33][34][35].
In case of fully laminar flow, due to the friction, low velocity occurs at the tube (vessel) wall, and the highest flow velocity can be observed along the axis, resulting in a parabolic velocity profile in the longitudinal cross-section of the tube. The tangent of the streamlines reflects the flow direction at a given point. The concentration of the streamlines determines the magnitude of the flow. At the vicinity of the tube wall the flow is dominantly laminar, while along the axis the velocity is higher and its homogeneity is smaller, with turbulences developing as a consequence. This is due to the unbalance between inertial and frictional forces when velocity increases. Turbulence starts above ~2100 Re (real turbulence: Re > 104). Real turbulence—by definition—does not occur in the vasculature. The critical velocity shows when the flow becomes turbulent: νcrit = Re ηρ [24][25][33].
The Δp versus Q curve is almost linear below 1000 Re (Q ~ Δp); above it the curve starts to flatten (Q ~ √Δp): when increasing the pressure-gradient the flow-increase does not increase in the same manner and, due to the developing turbulence, the flow resistance increases [36].
Further effects occurring include Dean-vortices and the Magnus-effect. In the circulation the pulsatility also takes part in forming the flow profile, showing a decreasing effect toward the capillaries. The Wommersley-number reflects the pulsatility: α = R (ωρ/η)1/2, where R: radius, ω: heart frequency ρ: blood density, η: blood viscosity. Pressure-oscillation also has to be mentioned here, which is composed of various frequency components: first-order (pulsating), second-order (respiration-related), third-order (Traube–Hering–Mayer) waves. The characteristics of blood flow are determined not only by the properties of the circulating blood, but also by the geometry of the vascular system, its regulatory mechanisms, and the passive biomechanics of the vessel wall.
Numerous endothelial functions are modulated by the shearing force, so by the rheological properties of the blood and the flow profile: membrane ion channel proteins, receptor-dependent G-proteins, protein kinase cascades, transcription factors, such as NF-κB, BGR-1, c-FOS, among others. Cytoskeleton, membrane caveolae, junctional proteins, adhesion complexes also take part in shear-induced endothelial responses. Fluid shear stress plays an important role in physiological and pathological vascular remodeling as well, via junctional mechanosensory complex (PECAM-1, VEGFRs, VE-Cad) and CCM complexes (CCM1, CCM2, CCM3). Consequently, any changes in blood rheology (increased blood and/or plasma viscosity, impaired red blood cell deformability, enhanced red blood cell aggregation) strongly affect endothelial functions [28][29][30][31][37].
2.2. Blood and Plasma Viscosity
The blood is a non-Newtonian, thixotropic and viscoelastic fluid. Its viscosity depends on the shear rate. The non-Newtonian characteristic of the blood is hematocrit and shear rate dependent. At lower shear rate the blood viscosity increases due to the viscosity elevating effect of the red blood cell aggregation. At higher flow velocity (i.e., higher shear rate), the cells disaggregate and elongate in the direction of the flow due to their deformability and elastic features [24][25][33]. The connection between blood viscosity and hematocrit (Htc) shows an exponential-like rather than a linear relation [24]. The hematocrit/viscosity rate presented in the function of the hematocrit shows a connection resembling of a bell-shaped curve, with a peak (by derivation of the curve), which reflects the maximum of oxygen transporting capacity of blood. It is concerned as an “optimal” hematocrit, i.e., the possible highest red blood cell count (the highest hematocrit) at the possible lowest viscosity [38].
Plasma viscosity is primarily determined by its water content and the macromolecules with elongated configuration, such as certain plasma proteins (fibrinogen, globulin fractions), besides triglycerides and lipoproteins [25]. Along the vasculature the apparent viscosity of the blood is not constant due to the intravascular interactions and distribution of different shear rate profile and the formed elements, mainly the red blood cells; however, the intravascular plasma viscosity can be considered constant. Consequently, the plasma viscosity maintains shear stress on the endothelium at the cell-poor or often cell-free zone in the direct vicinity of the endothelium (Poiseuille-zone), due to the axial flow profile in vessels with diameter approximately under 300 μm [25].
2.3. Red Blood Cell Deformability
The ability of passive deformation of the red blood cells by shearing and compressing forces depends on the absolute volume, surface/volume ratio of the cells, morphological characteristics, viscoelastic properties of the cell membrane and intracellular viscosity [24][25][39][40][41][42].
These features are dominantly derived from the molecular composition of the cell membrane, cytoskeletal structure. More than 50 transmembrane proteins are known in erythrocytes: transport proteins (anion transporter band-3, water transporter aquaporin-1, glucose and l-dehydroascorbinic acid transporter glut1, urea transporter Kidd antigen protein, transporter RhAG, Na+/K+-ATPase, Ca2+ATPase, Na+-K+-2Cl−, Na+-Cl−, Na+-K+, K+-Cl− co-transporters, Gárdos-channel), adhesion molecules, receptor, blood group antigens, A–D glycophorin complexes, and other proteins. This structure gives the red blood cells’ membrane elasticity and mechanical stability. From pathophysiological point of view, it is important to mention that this stability depends on the composition of the proteins, phosphorilation state, intracellular Ca2+ concentration, and free-radical reactions affect their structure and function. More than 340 membrane proteins are known. Their complexity is high, about 80% of them are still not associated with structural–functional models; their roles have not been fully elucidated yet [43][44].
Mammalian mature red blood cells are unnucleated, so the intracellular viscosity is mainly determined by the properties of hemoglobin content. Mean cell hemoglobin concentration is regulated within a relatively narrow range (30−35 g/d). It is also well-known that ATP-dependent Na+, K+ transporters participate in the cell volume regulation [23][25][39][44][45].
Viscosity property derives from membrane viscosity and cytoplasm viscosity, and elasticity characteristic is associated with the surface expansion, shearing and bending elements [46]. Motion of red blood cells depends on the flow conditions: rolling, rotation, swinging, elongation, elastic deformation, the combination of all the above [34][35][47]. Worsening deformability, i.e., the enhanced rigidity of red blood cells, in the mass flow zone of circulation (vessel diameter >300 μm) may lead to elevated blood viscosity. Red blood cells with reduced deformability can cause the most significant problem in the microcirculation, more precisely in the zone of so-called individual flow. Here, capillaries with diameter of 3–5 μm may also occur. Deformability is essential to passing through these capillaries.
2.4. Red Blood Cell Aggregation
Under stasis or at low shear rate erythrocytes reversibly clump together. Initially they arrange next to each other in a rouleaux formation, resembling a stack of coins. It happens within a few seconds (1–5 s). The rouleaux form larger two- then three-dimensional shape aggregates in the next seconds or even minutes [23]. In anticoagulated blood samples due to the gravity these aggregates show sedimentation. If the aggregation is fast and/or extensive, thus the erythrocyte sedimentation rate (ESR) values are higher.
Red blood cell aggregability is influenced by cellular factors. The aggregation happens when plasmatic factors are also presented. The process of red blood cell aggregation is not completely understood yet. There are two theories. (1) Bridging model: the aggregation happens via non-covalent cross-linking of macromolecules (large proteins with elongated structure, such as fibrinogen, or in vitro with different polymers). (2) Depletion model: since the glycocalyx layer does not allow the macromolecules to penetrate close to the membrane, a depletion zone develops in the vicinity of the cells. As two red blood cells approach each other, due to the osmotic gradient between the macromolecule concentration of the plasma phase and the depletion zone, a kind of “pulling” force may develop [23]. Disaggregating forces include electrostatic repulsion (due to surface negative charge), membrane strain, and shearing forces. There is no clear consensus about these two theories. Maybe both might be true. Carvalho et al. using atomic-force microscopy showed that the αIIbβ3 glycoprotein complexes of erythrocytes (similar exist on platelets’ surface) have lower affinity to link fibrinogen. It is supposed that the depletion forces are important to “pull” the cells close enough to each other, and so weak linking may happen [48].
Various plasma proteins and in vitro different polymers of different molecular size influence the aggregation [23]. Fibrinogen, C-reactive protein and immunoglobulin M facilitate and enhance red blood cell aggregation. Immunoglobulin G and haptoglobin show rather increasing effects, while transferrin, coeruloplasmin and albumin do not influence erythrocyte aggregation [23]. In vitro high molecular weight macromolecules and polymers (e.g., 60 or 73 kDa dextran, 360 kDa polyvinylpyrrolidon, 36 kDa polyethylen-glycol) promote red blood cell aggregation. Armstrong et al. found that enhanced red blood cell aggregation was observed if the hydrodynamic radius (Rh) of the macromolecules/polymers was larger than 4 nm. Molecules with smaller Rh did not influence the aggregation process [49].
Red blood cell aggregation depends on the hematocrit (Hct) as well. However, the relationship is not linear. The aggregation index, which increases by the extent of aggregation, expresses a nearly linear relation between ~20 and 40% Hct, between about 40 and 50% the slope of the curve is smaller, while at higher Hct the aggregation does not change significantly with further increase in the Hct [23]. For aggregation the biconcave cell morphology is crucial, since ovalocyte, spherocyte, echinocyte, sphero-echinocyte cell forms are less capable of aggregating [23][26]. Due to the complex background given by the cellular and plasmatic factors, red blood cell aggregation shows the highest diversity amongst animal species compared to human [23][26][50][51].
2.5. Hemorheological Factors in Microcirculation
Fåhræus observed in 1958 that by enhanced aggregation the axial flow in the glass capillary is more expressed and the cell-free side zone is wider [52]. The parabolic flow rate profile and the presence of circulating blood cells lead to the phenomenon that typically appears in the range under 200–300 µm vein diameter: the axial migration of red blood cells. Along the vessel’s cross section, the distribution and velocity of red blood cells lead to the dynamic reduction in hematocrit (Fåhræus-effect). Below approximately 30 µm diameter, down to about 10 µm, the apparent blood viscosity decreases with the diameter (Fåhræus–Lindqvist effect) [53]. Along the vessel wall the cell-poor/cell-free Poiseuille-zone reduces the friction, and thereby the hydrodynamic resistance. When the shear-rate enables the development of red blood cell aggregation (typically in the area of postcapillary venules), the aggregates also flow along the axis. Increased aggregation enlarges the circulating particles, so enhancing the axial migration. Consequently, the Poiseuille-zone widens which process facilitates the margination of leukocytes [54][55] (Figure 3).
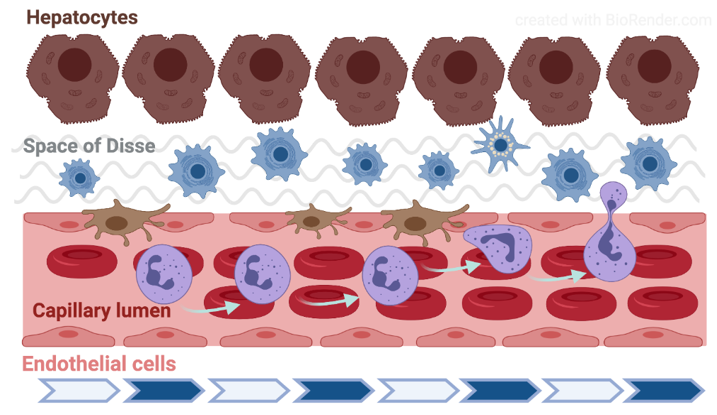
Figure 3. Schematic drawing of the liver microcirculation. The figure was created with BioRender (https://biorender.com).
In the microcirculation due to the extremely variable ramifications, connections, bends, endothelial surface features, red blood cell distribution, the tissue hematocrit is highly variable [34][35][56][57][58].
3. Characteristics of the liver circulation
3.1. Circulation and microcirculation
The liver receives its blood supply both through the portal vein (around 75%) and the hepatic artery. These two vessels enter the liver and penetrate the liver parenchyma together and in close vicinity, and gradually branch until arterioles and venules. Upon reaching the periphery of the hexagonal liver lobules separately, the terminal portal venules enter the lobule in-between the cords of hepatocytes and continue with the sinusoids, a microvascular network which forms most of the capillary bed in the hepatic parenchyma [59][60][61]. The hepatic artery supplies oxygenated blood to the biliary tree (through the peribiliary plexus), portal tract interstitium and the vasa vasorum of the intrahepatic vessels. The arterioles also ultimately empty in the sinusoids resulting in mixing of arterial and portal blood. The sinusoids run straight for about 250 µm and converge radially towards the center of the lobule where all empty into the central vein. The sinusoids communicate with each other through shorter interconnecting sinusoids running across the liver cell and their diameter gradually increase from 5 to 7 µm in the periphery (portal zone) to 10 to 15 µm in the pericentral area [62] (Figure 3).
The ultrastructure of sinusoidal endothelial cells (SEC) differs greatly from other capillaries in the body due to the presence of sieve-like pores (or fenestrae) measuring between 100 and 200 nm and by the lack of a basement membrane [63]. These unique features result in a high, bidirectional permeability of the sinusoids to macromolecules, solutes and water. Besides the SEC, the microvascular network contains four other distinct components: the hepatic stellate cells (HSC), which are located in the space of Disse and modulate sinusoidal tone and stiffness; the Kupffer cells (KC), unique liver-resident macrophages anchored to the luminal site of the endothelium and exposed to the bloodstream; the hepatocytes, the parenchymal cells that are tightly attached to SECs and account for the liver metabolism; and the extracellular matrix (ECM), the scaffold of the entire structure, which serves as a niche for these cells and may influence their phenotype depending on its composition. SECs and KCs belong to the mononuclear phagocyte system (MPS), also known as the reticuloendothelial system. The majority of KCs are found in the peripheral, periportal area where they are larger and have greater phagocytic activity than those located in the perilobular region [62]. Both SECs and KCs are very sensitive to ischemia-reperfusion injury and are key elements in the local response after liver ischemia. Thus, the sinusoidal cells become swollen and may detach already during ischemia whereas KCs are activated during the early phase after reperfusion releasing free radicals, vasoactive mediators and proinflammatory cytokines in the ischemic areas [64][65]. The hepatic stellate cells reside outside the sinusoid, between the basal surfaces of the hepatocytes and the SEC, in the space of Disse [66]. In contrast to the KCs, HSCs are distributed homogeneously throughout the three zones of the liver lobule. Their finger-like projections called perisinusoidal processes surround the sinusoidal tube and may regulate its tone. HSCs produce various inflammatory molecules, interact with other liver cells and relay and integrate the signals from the sinusoids to the liver parenchyma. Hence, several cells may alter the delicate balance that maintains the microcirculatory homeostasis in the liver.
3.2. Rheological differences (aorto-porto-caval)
There is only limited information about the characteristics of micro-rheological parameters in different vascular segments [26][67]. It has been demonstrated that red blood cell aggregation and deformability show significant aorto–porto–caval differences in rats [68]. The smallest elongation index values were found in the arterial samples, the highest values were in the systemic venous blood and the values of the portal blood sample fall between the two. Son et al. also found in rats that elongation index values are lower in arterial blood compared to venous blood. These differences could not be observed in human or in canine blood [67]. The aggregation index parameters are significantly lower in the systemic venous blood, than in the arterial and portal samples [68]. The explanation of these arterio–venous or porta–caval micro-rheological heterogeneity might be derived from the dynamic differences in pH, oxygenation level, lactate concentration, hematocrit and mean cell volume [22][23][26][40][69].
4. Pathophysiology of hemorheological alterations related to hepatic ischemia-reperfusion
During IRI, metabolic alterations (decrease in pH, increase in H+ and lactate) and osmolarity changes affect the morphological and mechanical properties of blood cells [40][69][70][71]. A decrease in pH will transform the normally discocyte-shape red bloods cells into stomacyte or sphero-stomacyte morphology. If the ATP depletion and calcium accumulation are the dominant effects, the echinocyte and sphero-echinocyte morphologies appear. Red blood cells’ deformability is reduced, and their aggregation is disturbed in both morphological transformations [26][40][69][70]. Change in oxygenation is also known to alter micro-rheology, since deformability of deoxygenated red blood cells is impaired, being associated with enhanced aggregation. Under hypoxia, the cell swelling alters surface to volume ratio, and consequently cellular deformability decreases.
Conditions and circumstances that lead to IRI (e.g., clamping vessels, obturation/occlusion, intravascular devices) cause mechanical trauma to blood [22][26][72]. Blood cells are continuously exposed to mechanical stress during their entire life-span in the circulation. A shear stress ranging around 5–20 Pa may even improve their deformability by releasing NO from the cells, besides other vasoactive mediators [26][51][73][74][75][76][77]. Higher shear stress, correlating with the magnitude and exposure time, causes mechanical trauma to the blood cells from sublethal trauma (decreased deformability, enhanced aggregation) and microvesicle generation to mechanical hemolysis [51][72][78].
Free radicals can be generated during reperfusion of a previously damaged tissue by ischemia (xanthine oxidase/reductase, neutrophils, NO synthetase activity when forming OONO− in the presence of NO and superoxide anion), and during the associated inflammatory reaction (neutrophils) [9][79]. Oxygen-centered free radicals can jeopardize red blood cells in several ways: lipid peroxidation on the cell membrane, sulfhydryl cross-linking and so altering structure and function of proteins (receptors, ion pumps, structural proteins), forming methemoglobin and Heinz-bodies. Red blood cells are very vulnerable for oxidative stress, because they are rich in iron (Fenton-reaction) and they do not have a nucleus; thus, they have no chance for new protein generation for any repair [9][22][27].
Due to inflammatory processes the developing acute phase reactions also cause non-specific hemorheological changes: elevated fibrinogen and α2-macroglobulin concentration accompanied by consequent increase in plasma viscosity, rise in leukocyte count, increase or decrease in platelet count, hemoconcentration, besides micro-rheological alterations [9][22][27]. Enhanced red blood cell aggregation elevates blood viscosity and increases the flow resistance: the axial migration of the cells becomes more expressed, widening Poiseuille-zone, facilitated leukocyte tethering and margination, slowed down rolling are also included [55].
Hypoxia leads to impaired endothelial cell barrier function and, additionally, altered blood rheology has an impact on the shear stress profile on the endothelial surface modulating numerous functions as discussed before. If hypovolemia is also associated with ischemia-reperfusion, due to the increased sympathetic activation vasoconstriction appears, leading to reduction in the capillary cross-sectional area and endothelial swelling. In the microcirculatory bed, the “no-reflow” phenomenon is characteristic for ischemia-reperfusion that is caused by microvascular spasm, swelling of endothelial cells, bleb formation on the endothelial surface, increased capillary permeability, interstitial edema, micro-thrombi, neutrophil adhesion and plugging, local acidosis, and presence of red blood cells with altered micro-rheology (impaired deformability and enhanced aggregation) [27][62][80][81][82][83][84][85]. Hemorheological and microcirculatory alterations in ischemia-reperfusion may show age- and gender-related differences as well [75][86][87].
In summary, the initiating effects (tissue damage, hypoxia, mechanical trauma to blood, free radicals, and their combinations) lead to metabolic alterations and inflammatory processes affecting micro- and macro-rheological parameters, flow characteristics and endothelial functions, resulting in microcirculatory disturbances and decrease in perfusion. These generate further tissue damage.
Effect of Pringle/Baron Maneuver
Despite of the availability of modern devices controlling bleeding of parenchymal organs, temporarily clamping the hepatoduodenal ligament (Baron or Pringle maneuver) is still widely used in liver surgery for hepatic blood inflow control during major resections (open and laparoscopic/robotic surgery) to reduce bleeding and the need for perioperative blood transfusion [88][89][90][91][92]. Despite its obvious technical benefits during surgery, its use remains controversial due to potential negative effects on the clinical outcomes. Thanks to modern anesthesiology, intensive care, surgical devices and methods, the ischemic time tolerated by the liver has been extended in the past decades. Prolonged vascular inflow occlusion (≥60 min) can be safely applied using both continuous and intermittent regimens [93]. When performing the Pringle maneuver, besides hepatic ischemia, venous congestion also appears in the portal system, causing duration-dependent damage to the intestines as well [94].
Concerning hemorheological and microcirculatory changes, ischemia and reperfusion, congestion, hemoconcentration, metabolic alterations, all are important. In a canine model it has been demonstrated that the intermittent Pringle maneuver (three times, 15-min of ischemia alternating with 5-min of reperfusion) caused increased hematocrit and red blood cell aggregation in the systemic and hepatic venous blood samples immediately after the first maneuver. Followed by the second clamping, this increase was not seen, while after the third maneuver, red blood cell aggregation was enhanced again not only in the systemic, but also in the portal venous blood together with an increase in local hematocrit values [95].
5. Conclusions
In conclusion, hemorheological factors play an important role determining tissue perfusion and orchestrate mechanical stress-dependent endothelial functions. During IRI dominantly non-specific alterations lead to deteriorating micro-rheological properties as well. Antioxidant and anti-inflammatory agents, ischemic conditioning protocols and machine perfusion may improve rheological properties in the hepatic sinusoids contributing to the protection against hepatic IRI. Studying hemorheological factors together with microcirculatory investigations may provide useful information for better understating the pathomechanism of hepatic ischemia-reperfusion and to optimize surgical conditioning protocols and organ preservation techniques.
References
- Starzl, T.E.; Fung, J.J. Themes of liver transplantation. Hepatology 2010, 51, 1869–1884.
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Liver transplantation. J. Hepatol. 2016, 64, 433–485.
- Nemes, B.; Gaman, G.; Polak, W.G.; Gelley, F.; Hara, T.; Ono, S.; Baimakhanov, Z.; Piros, L.; Eguchi, S. Extended-criteria donors in liver transplantation Part II: Reviewing the impact of extended-criteria donors on the complications and outcomes of liver transplantation. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 841–859.
- Czigany, Z.; Lurje, I.; Schmelzle, M.; Schöning, W.; Öllinger, R.; Raschzok, N.; Sauer, I.M.; Tacke, F.; Strnad, P.; Trautwein, C.; et al. Ischemia-reperfusion injury in marginal liver grafts and the role of hypothermic machine perfusion: Molecular mechanisms and clinical implications. J. Clin. Med. 2020, 9, 846.
- Szijarto, A.; Czigany, Z.; Turoczi, Z.; Harsanyi, L. Remote ischemic preconditioning-a simple, low-risk method to decrease ischemic reperfusion injury: Models, protocols and mechanistic background. A review. J. Surg. Res. 2012, 178, 797–806.
- Czigany, Z.; Lurje, I.; Tolba, R.H.; Neumann, U.P.; Tacke, F.; Lurje, G. Machine perfusion for liver transplantation in the era of marginal organs-New kids on the block. Liver Int. 2018, 39, 228–249.
- Nakamura, K.; Kageyama, S.; Kupiec-Weglinski, J.W. Innate immunity in ischemia-reperfusion injury and graft rejection. Curr. Opin. Organ. Transplant. 2019, 24, 687–693.
- Li, J.; Li, R.J.; Lv, G.Y.; Liu, H.Q. The mechanisms and strategies to protect from hepatic ischemia-reperfusion injury. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 2036–2047.
- Nemeth, N.; Toth, E.; Nemes, B. Agents targeting ischemia-reperfusion injury. In Current Immunosuppressive Therapy in Organ Transplantation; Huifang, C., Shiguang, Q., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2015; pp. 487–533.
- Yang, W.; Chen, J.; Meng, Y.; Chen, Z.; Yang, J. Novel targets for treating ischemia-reperfusion injury in the liver. Int. J. Mol. Sci. 2018, 19, 1302.
- Abu-Amara, M.; Yang, S.Y.; Tapuria, N.; Fuller, B.; Davidson, B.; Seifalian, A. Liver ischemia/reperfusion injury: Processes in inflammatory networks—A review. Liver Transplant. 2010, 16, 1016–1032.
- Perry, B.C.; Soltys, D.; Toledo, A.H.; Toledo-Pereyra, L.H. Tumor necrosis factor-alpha in liver ischemia/reperfusion injury. J. Investig. Surg. 2011, 24, 178–188.
- Peralta, C.; Jimenez-Castro, M.B.; Gracia-Sancho, J. Hepatic ischemia and reperfusion injury: Effects on the liver sinusoidal milieu. J. Hepatol. 2013, 59, 1094–1106.
- Kageyama, S.; Hata, K.; Tanaka, H.; Hirao, H.; Kubota, T.; Okamura, Y.; Iwaisako, K.; Takada, Y.; Uemoto, S. Intestinal ischemic preconditioning ameliorates hepatic ischemia/reperfusion injury in rats: Role of heme oxygenase 1 in the second window of protection. Liver Transplant. 2015, 21, 112–122.
- Lu, L.; Zhou, H.; Ni, M.; Whang, X.; Busuttil, R.; Kupiec-Weglinski, J.; Zhai, Y. Innate immune regulations and liver ischemia-reperfusion injury. Transplantation 2016, 100, 2601–2610.
- Covington, S.M.; Bauler, L.D.; Toledo-Pereyra, L.H. Akt: A therapeutic target in hepatic ischemia–reperfusion injury. J. Investig. Surg. 2017, 30, 47–55.
- Li, Y.; Ma, D.; Wang, Z.; Yang, J. MicroRNA-155 Deficiency in Kupffer cells ameliorates liver ischemia-reperfusion injury in mice. Transplantation 2017, 101, 1600–1608.
- Zimmerman, M.A.; Martin, A.; Yee, J.; Schiller, J.; Hong, J.C. Natural killer T cells in liver ischemia-reperfusion injury. J. Clin. Med. 2017, 6, 41.
- Oliveira, T.H.C.; Marques, P.E.; Proost, P.; Teixeira, M.M.M. Neutrophils: A cornerstone of liver ischemia and reperfusion injury. Lab. Investig. 2018, 98, 51–62.
- Dar, W.A.; Sullivan, E.; Bynon, J.S.; Eltzschig, H.; Ju, C. Ischaemia reperfusion injury in liver transplantation: Cellular and molecular mechanisms. Liver Int. 2019, 39, 788–801.
- Nakamura, K.; Kageyama, S.; Kupiec-Weglinski, J.W. The evolving role of neutrophils in liver transplant ischemia-reperfusion injury. Curr. Transplant. Rep. 2019, 6, 78–89.
- Baskurt, O.K. Mechanisms of blood rheology alterations. In Handbook of Hemorheology and Hemodynamics; Baskurt, O.K., Hardeman, M.R., Rampling, M.W., Meiselman, H.J., Eds.; IOS Press: Amsterdam, The Netherlands, 2007; pp. 170–190.
- Baskurt, O.K.; Neu, B.; Meiselman, H.J. Determinants of red blood cell aggregation. In Red Blood Cell Aggregation; Baskurt, O.K., Neu, B., Meiselman, H.J., Eds.; CRC Press: Boca Raton, FL, USA, 2012; pp. 9–29.
- Cokelet, G.R.; Meiselman, H.J. Basic aspects of hemorheology. In Handbook of Hemorheology and Hemodynamics; Baskurt, O.K., Hardeman, M.R., Rampling, M.W., Meiselman, H.J., Eds.; IOS Press: Amsterdam, The Netherlands, 2007; pp. 21–33.
- Cokelet, G.R.; Meiselman, H.J. Macro- and micro-rheological properties of blood. In Handbook of Hemorheology and Hemodynamics; Baskurt, O.K., Hardeman, M.R., Rampling, M.W., Meiselman, H.J., Eds.; IOS Press: Amsterdam, The Netherlands, 2007; pp. 45–71.
- Nemeth, N.; Deak, A.; Szentkereszty, Z.; Peto, K. Effects and influencing factors on hemorheological variables taken into consideration in surgical pathophysiology research. Clin. Hemorheol. Microcirc. 2018, 69, 133–140.
- Nemeth, N.; Szabo, A. Microcirculation. In Advances in Experimental Surgery; Huifang, C., Martins, P., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2018; Volume 2, pp. 317–357.
- Weinbaum, S.; Zhang, X.; Han, Y.; Vink, H.; Cowin, S.C. Mechanotransduction and flow accross the endothelial glycocalyx. PNAS 2003, 100, 7988–7995.
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Investig. 2016, 126, 821–828.
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2017, 219, 382–408.
- Souilhol, C.; Serbanovic-Canic, J.; Fragiadaki, M.; Chico, T.J.; Ridger, V.; Roddie, H.; Evans, P.C. Endothelial responses to shear stress in atherosclerosis: A novel role for developmental genes. Nat. Rev. Cardiol. 2020, 17, 52–63.
- Copley, A.L. 1985: History of clinical hemorheology. Clin. Hemorheol. 1985, 5, 765–812.
- Chandran, K.B.; Rittgers, S.E.; Yoganathan, A.P. (Eds.) Rheology of blood and vascular mechanics. In Biofluid Mechanics; CRC Press: Boca Raton, FL, USA, 2012; pp. 109–154.
- Lipowsky, H.H. Shear stress in the circulation. In Flow Dependent Regulation of Vascular Function; Clinical Physiology Series; Bevan, J., Kaley, G., Eds.; Oxford University Press: New York, NY, USA, 1995; pp. 28–45.
- Lipowsky, H.H. Microvascular rheology and hemodynamics. Microcirculation 2005, 12, 5–15.
- Monos, E. Hemodynamics. Biomechanics of the Blood Circulation; Semmelweis Publishers: Budapest, Hungary, 2011.
- Fernández-Ruiz, I. Plexin D1 is a mechanosensor that regulates the site-specific distribution of atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 199.
- Piety, N.Z.; Reinhart, W.H.; Stutz, J.; Shevkoplyas, S.S. Optimal hematocrit in an artificial microvascular network. Transfusion 2017, 57, 2257–2266.
- Chien, S. Biophysical behavior of red cells in suspension. In The Red Blood Cell; Surgenor, D.M., Ed.; Academic Press: New York, NY, USA, 1975; pp. 1031–1133.
- Meiselman, H.J. Morphological determinants of red blood cell deformability. Scand. J. Clin. Lab. Investig. 1981, 41 (Suppl. 156), 27–34.
- De Oliveira, S.; Saldanha, C. An overview about erythrocyte membrane. Clin. Hemorheol. Microcirc. 2010, 44, 63–74.
- Huisjes, R.; Bogdanova, A.; van Solinge, W.W.; Schiffelers, R.M.; Kaestner, L.; van Wijk, R. Squeezing for life-properties of red blood cell deformability. Front. Physiol. 2018, 9, 656.
- Mohandas, N.; Chasis, J.A. Red blood cell deformability, membrane material properties and shape: Regulation by transmembrane, skeletal and cytosolic proteins and lipids. Semin. Hematol. 1993, 30, 171–192.
- Mohandas, N.; Gallagher, P.G. Red cell membrane, past, present, and future. Blood 2008, 112, 3939–3948.
- Pretini, V.; Koenen, M.H.; Kaestner, L.; Fens, M.H.A.M.; Schiffelers, R.M.; Bartels, M.; Van Wijk, R. Red blood cells: Chasing interactions. Front. Physiol. 2019, 10, 945.
- Thurston, G.B.; Henderson, N.M. Viscoelasticity of human blood. In Handbook of Hemorheology and Hemodynamics; Baskurt, O.K., Hardeman, M.R., Rampling, M.W., Meiselman, H.J., Eds.; IOS Press: Amsterdam, The Netherlands, 2007; pp. 72–90.
- Bitbol, M. Red blood cell orientation in orbit C = 0. Biophys. J. 1986, 49, 1055–1068.
- Carvalho, F.A.; Connell, S.; Miltenberger-Miltenyi, G.; Pereira, S.V.; Tavares, A.; Ariëns, R.A.; Santos, N.C. Atomic force microscopy-based molecular recognition of a fibrinogen receptor on human erythrocytes. ACS Nano 2010, 4, 4609–4620.
- Armstrong, J.K.; Wenby, R.B.; Meiselman, H.J.; Fisher, T.C. The hydrodynamic radii of macromolecules and their effect on red blood cell aggregation. Biophys. J. 2004, 87, 4259–4270.
- Windberger, U.; Baskurt, O.K. Comparative hemorheology. In Handbook of Hemorheology and Hemodynamics; Baskurt, O.K., Hardeman, M.R., Rampling, M.W., Meiselman, H.J., Eds.; IOS Press: Amsterdam, The Netherlands, 2007; pp. 267–285.
- Nemeth, N.; Sogor, V.; Kiss, F.; Ulker, P. Interspecies diversity of erythrocyte mechanical stability at various combinations in magnitude and duration of shear stress, and osmolality. Clin. Hemorheol. Microcirc. 2016, 63, 381–398.
- Fåhraeus, R. The influence of the rouleau formation of the erythrocytes on the rheology of the blood. Acta Med. Scand. 1958, 161, 151–165.
- Fåhraeus, R.; Lindqvist, T. The viscosity of the blood in narrow capillary tubes. Am. J. Physiol. 1931, 96, 562–568.
- Baskurt, O.K. In vivo correlates of altered blood rheology. Biorheology 2008, 45, 629–638.
- Nash, G.B.; Watts, T.; Thornton, C.; Barigou, M. Red cell aggregation as a factor influencing margination and adhesion of leukocytes and platelets. Clin. Hemorheol. Microcirc. 2008, 39, 303–310.
- Maeda, N. Erythrocyte rheology in microcirculation. Jpn. J. Physiol. 1996, 46, 1–14.
- Pries, A.R.; Secomb, T.W. Rheology of the microcirculation. Clin. Hemorheol. Microcirc. 2003, 29, 143–148.
- Pries, A.R.; Secomb, T.W. Blood flow in microvascular networks. In Handbook of Physiology, Microcirculation, 2nd ed.; Tuma, R.F., Duran, W.N., Ley, K., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2008; pp. 3–36.
- Ekataksin, W.; Kaneda, K. Liver microvascular architecture: An insight into the pathophysiology of portal hypertension. Semin. Liver Dis. 1999, 19, 359–382.
- Miura, S.; Kubes, P.; Granger, D.N. Gastrointestinal and liver microcirculations: Roles in inflammation and immunity. In Handbook of Physiology, Microcirculation, 2nd ed.; Tuma, R.F., Duran, W.N., Ley, K., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2008; pp. 684–711.
- Gracia-Sancho, J.; Marrone, G.; Fernández-Iglesias, A. Hepatic microcirculation and mechanisms of portal hypertension. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 221–234.
- Vollmar, B.; Menger, M.D. The hepatic microcirculation: Mechanistic contributions and therapeutic targets in liver injury and repair. Physiol. Rev. 2009, 89, 1269–1339.
- Braet, F.; Wisse, E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp. Hepatol. 2002, 1, 1.
- DeLeve, L.D. Hepatic microvasculature in liver injury. Semin. Liver Dis. 2007, 27, 390–400.
- Brunt, E.M.; Gouw, A.S.; Hubscher, S.G.; Tiniakos, D.G.; Bedossa, P.; Burt, A.D.; Callea, F.; Clouston, A.D.; Dienes, H.P.; Goodman, Z.D.; et al. Pathology of the liver sinusoids. Histopathology 2014, 64, 907–920.
- McCuskey, R.S. Morphological mechanisms for regulating blood flow through hepatic sinusoids. Liver 2000, 20, 3–7.
- Son, K.H.; Lim, C.H.; Song, E.J.; Sun, K.; Son, H.S.; Lee, S.H. Inter-species hemorheologic differences in arterial and venous blood. Clin. Hemorheol. Microcirc. 2010, 44, 27–33.
- Klarik, Z.; Kiss, F.; Miko, I.; Nemeth, N. Aorto-porto-caval micro-rheological differences of red blood cells in laboratory rats: Further deformability and ektacytometrial osmoscan data. Clin. Hemorheol. Microcirc. 2013, 53, 217–229.
- Reinhart, W.H.; Gaudenz, R.; Walter, R. Acidosis induced by lactate, pyruvate, or HCl increases blood viscosity. J. Crit. Care 2002, 17, 38–42.
- Reinhart, W.H.; Chien, S. Red cell rheology in stomatocyte-echinocyte transformation, roles of cell geometry and cell shape. Blood 1980, 67, 1110–1118.
- Reinhart, W.H.; Piety, N.Z.; Goede, J.S.; Shevkoplyas, S.S. Effect of osmolality on erythrocyte rheology and perfusion of an artificial microvascular network. Microvasc. Res. 2015, 98, 102–107.
- Kameneva, M.V.; Antaki, J.F. Mechanical trauma to blood. In Handbook of Hemorheology and Hemodynamics; Baskurt, O.K., Hardeman, M.R., Rampling, M.W., Meiselman, H.J., Eds.; IOS Press: Amsterdam, The Netherlands, 2007; pp. 206–227.
- Cortese-Krott, M.M.; Kelm, M. Endothelial nitric oxide synthase in red blood cells: Key to a new erythrocrine function? Redox. Biol. 2014, 2, 251–258.
- Diederich, L.; Suvorava, T.; Sansone, R.; Keller, T.C.S., 4th; Barbarino, F.; Sutton, T.R.; Kramer, C.M.; Lückstädt, W.; Isakson, B.E.; Gohlke, H.; et al. On the effects of reactive oxygen species and nitric oxide on red blood cell deformability. Front. Physiol. 2018, 9, 332.
- Grau, M.; Cremer, J.M.; Schmeichel, S.; Kunkel, M.; Bloch, W. Comparisons of blood parameters, red blood cell deformability and circulating nitric oxide between males and females considering hormonal contraception: A longitudinal gender study. Front. Physiol. 2018, 9, 1835.
- McMahon, T.J. Red blood cell deformability, vasoactive mediators, and adhesion. Front. Physiol. 2019, 10, 1417.
- Semenov, A.N.; Shirshin, E.A.; Muravyov, A.V.; Priezzhev, A.V. The effects of different signaling pathways in adenylyl cyclase stimulation on red blood cells deformability. Front. Physiol. 2019, 10, 923.
- Leal, J.K.F.; Adjobo-Hermans, M.J.W.; Bosman, G.J.C.G.M. Red blood cell homeostasis: Mechanisms and effects of microvesicle generation in health and disease. Front. Physiol. 2018, 9, 703.
- Eltzschig, H.; Eckle, T. Ischemia and reperfusion–from mechanism to translation. Nat. Med. 2011, 17, 1391–1401.
- Granger, D.N. Physiology and pathophysiology of the microcirculation. Dial. Cardiovasc. Med. 1988, 3, 123–140.
- Menger, M.D.; Steiner, D.; Messmer, K. Microvascular ischemia-reperfusion injury in striated muscle, significance of ‘no-reflow’. Am. J. Physiol. 1992, 263, H1892–H1900.
- Reffelmann, T.; Kloner, R.A. The “no-reflow” phenomenon, basic science and clinical correlates. Heart 2002, 87, 162–168.
- Vollmar, B.; Menger, M. Intestinal ischemia/reperfusion, microcirculatory pathology and functional consequences. Langenbecks Arch. Surg. 2011, 396, 13–29.
- Yu, H.; Kalogeris, T.; Korthuis, R.J. Reactive species-induced microvascular dysfunction in ischemia/reperfusion. Free Radic. Biol. Med. 2019, 135, 182–197.
- Guven, G.; Hilty, M.P.; Ince, C. Microcirculation: Physiology, pathophysiology, and clinical application. Blood Purif. 2020, 49, 143–150.
- Maeso-Diaz, R.; Ortega-Ribeira, M.; Fernandez-Iglesias, A.; Hide, D.; Munoz, L.; Hessheimer, A.J.; Vila, S.; Frances, R.; Fondevila, C.; Albillos, A.; et al. Effects of aging on liver microcirculatory function and sinusoidal phenotype. Aging Cell 2018, 17, e12829.
- Mester, A.; Magyar, Z.; Molnár, Á.; Somogyi, V.; Tánczos, B.; Pető, K.; Németh, N. Age- and gender-related hemorheological alterations in intestinal ischemia-reperfusion in the rat. J. Surg. Res. 2018, 225, 68–75.
- Gurusamy, K.S.; Sheth, H.; Kumar, Y.; Sharma, D.; Davidson, B.R. Methods of vascular occlusion for elective liver resections. Cochrane Database Syst. Rev. 2009, 1, CD007632.
- Gurusamy, K.S.; Gonzalez, H.D.; Davidson, B.R. Current protective strategies in liver surgery. World J. Gastroenterol. 2010, 16, 6098–6103.
- Piardi, T.; Lhuaire, M.; Memeo, R.; Pessaux, P.; Kianmanesh, R.; Sommacale, D. Laparoscopic Pringle maneuver: How we do it? Hepatobiliary Surg. Nutr. 2016, 5, 345–349.
- Wei, X.; Zheng, W.; Yang, Z.; Liu, H.; Tang, T.; Li, X.; Liu, X. Effect of the intermittent Pringle maneuver on liver damage after hepatectomy: A retrospective cohort study. World J. Surg. Oncol. 2019, 17, 142.
- Al-Saeedi, M.; Ghamarnejad, O.; Khajeh, E.; Shafiei, S.; Salehpour, R.; Golriz, M.; Mieth, M.; Weiss, K.H.; Longerich, T.; Hoffmann, K.; et al. Pringle maneuver in extended liver resection: A propensity score analysis. Sci. Rep. 2020, 10, 8847.
- Van Riel, W.G.; van Golen, R.F.; Reiniers, M.J.; Heger, M.; van Gulik, T.M. How much ischemia can the liver tolerate during resection? Hepatobiliary Surg. Nutr. 2016, 5, 58–71.
- Dello, S.A.; Reisinger, K.W.; van Dam, R.M.; Bemelmans, M.H.; van Kuppevelt, T.H.; van den Broek, M.A.; Olde Damink, S.W.; Poeze, M.; Buurman, W.A.; Dejong, C.H. Total intermittent Pringle maneuver during liver resection can induce intestinal epithelial cell damage and endotoxemia. PLoS ONE 2012, 7, e30539.
- Furka, A.; Nemeth, N.; Gulyas, A.; Brath, E.; Peto, K.; Takacs, I.E.; Furka, I.; Sapy, P.; Miko, I. Hemorheological changes caused by intermittent Pringle (Baron) maneuver in beagle canine model. Clin. Hemorheol. Microcirc. 2008, 40, 177–189.

