 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ilias Mylonis | + 2026 word(s) | 2026 | 2021-02-20 04:30:54 | | | |
| 2 | Camila Xu | + 6 word(s) | 2032 | 2021-03-17 06:41:30 | | |
Video Upload Options
The Hypoxia Inducible Factor (HIF) family of heterodimeric transcription factors that consists of 3 HIFα members (namely HIF-1α, HIF-2α, and HIF-3α) and one HIFβ member (HIF-1β, best known as ARNT) is responsible for the transcriptional response of cells to oxygen deprivation.
1. The Hypoxia Inducible Factor (HIF) Family
The Hypoxia Inducible Factor (HIF) family of heterodimeric transcription factors consists of 3 HIFα members (namely HIF-1α, HIF-2α, and HIF-3α) and one HIFβ member (HIF-1β, best known as ARNT) [1]. HIF-1α is the most well studied member of the family and the first to be discovered by Semenza and coworkers [2][3][4] by its ability to bind to a hypoxia response element (HRE) in the 3′ enhancer of the human EPO gene. Unlike HIF-1α that can be expressed in all types of cells, HIF-2α, encoded by the EPAS1 gene, is expressed in a few tissues such as placenta, lungs, liver, and heart, and holds a central role in angiogenesis and erythropoiesis [5][6]. HIF-3α, the less studied isoform, has many spliced variants with distinct expression pattern [7] and diverging functionalities ranging from HIF-1 inhibition to transcriptional activation of HIF targets [8][9].
Detailed studies have shown that HIF-1α, as well as the other HIFα forms, are stabilized under hypoxia by an oxygen dependent mechanism (Figure 1) [10]. ARNT, which is constitutively expressed in cells regardless of oxygen concentration, associates with the HIFα subunits within the nucleus to form a functional heterodimer (HIF) that can bind to the HREs (Hypoxia Response Elements) of hypoxia target genes and initiate the transcriptional hypoxic response [11]. Heterodimerization and DNA binding are mediated by the Per-Arnt-Sim (PAS) homology and basic helix-loop-helix (bHLH) domains, respectively, which are present at the N-terminal parts of all HIF subunits [11]. Structural data indicate that both HIF isoforms bind HRE sequences in an identical fashion. The α-helices of their bHLH domains associate with the major groove of the recognition motif and their PAS-A domains cooperate with the bHLH domains of the heterodimer to establish binding to DNA [12]. Furthermore, the PAS domains of both HIFα isoforms possess cavities, which can accommodate small ligands, but their size and distribution differs between isoforms [12][13].
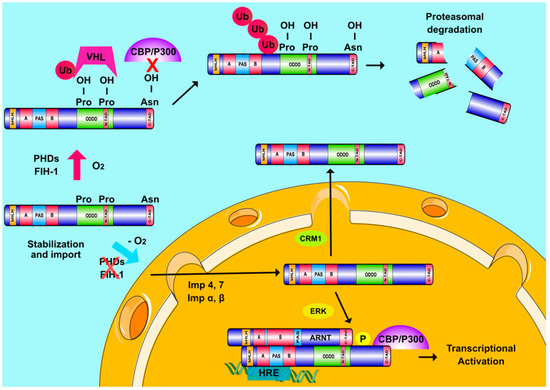
Figure 1. Regulation of HIFα subunits and Hypoxia Inducible Factors (HIF) transcriptional activity. When oxygen is abundant, PHDs and FIH hydroxylate two proline and one asparagine residue inside the ODDD and C-TAD regions of HIFα, respectively. Prolyl-hydroxylation leads to pVHL-mediated ubiquitination of HIFα subunits and their destruction in the proteasome while asparaginyl hydroxylation also inhibits HIFα interaction with CBP/P300. When oxygen levels drop, hydroxylases become inactive and HIFα subunits are stabilized and transported into the nucleus with the help of multiple importins (Imp α, β, 4, and 7). ERK-mediated phosphorylation of HIFα subunits ensures their nuclear accumulation by abrogating the association of HIFα with exportin CRM1. Nuclear HIFα subunits form a functional complex with ARNT, which binds to HREs on DNA and coactivators, such as CBP/p300 to induce transcription of hypoxia-regulated genes.
The C-terminal parts of both HIF-1α and HIF-2α are also critical for their function as they contain their oxygen dependent degradation domain (ODDD) as well as two distinct transactivation domains, N-TAD (overlapping with ODDD) and C-TAD (at the very C-terminus), which is responsible for the interaction between HIFα and the transcriptional coactivator proteins CBP/p300 (Figure 1) [14]. HIF-1 and HIF-2 activate a great number of genes (more than 1000) which can either be specific for each factor or common for both of them [15][1]. Domain-swapping experiments have suggested that HIF target gene specificity may be conferred by the N-TAD through its interaction with additional transcriptional co-regulators [16].
2. HIFs and Cellular Oxygen Sensing
The cellular oxygen sensing mechanism has been characterized in the previous decade mainly by the work of G. Semenza, Sir P. Ratcliffe and W. Kaelin (2019 Nobel prize in Physiology or Medicine). Their breakthrough experiments revealed that the HIFα subunits are subjected to hydroxylation in two specific prolyl residues when cells are grown under atmospheric oxygen concentrations (normoxia) [14]. This post-translational modification is essential for interaction with the Von Hippel-Lindau (pVHL) tumor suppressor protein that is part of a ubiquitin E3 ligase complex. As a result of this interaction, HIFα subunits are polyubiquitinated and targeted to the proteasome for destruction [17][18][19]. The enzymes catalyzing this hydroxylation are prolyl-hydroxylases PHD1, 2, and 3 in humans, also known as EGLN2,1 and 3. PHDs belong to the 2-oxoglutarate-dependent dioxygenase family and are thought to act as oxygen sensors since they use molecular oxygen as substrate. PHD2 is the most abundant and best studied isoform in cells [20][21][22][23]. A second oxygen dependent hydroxylation occurs at an asparagine residue in the C-TAD of HIFα and is catalyzed by a different oxygenase, called FIH (Factor Inhibiting HIF) [24][25]. FIH downregulates HIF transcriptional activity by impairing HIF binding to CBP/p300. According to all the above, HIFα subunits are constitutively produced but in the presence of normal oxygen concentrations both their expression and activity remain minimal. Under hypoxia, low O2 levels as well as the production of ROS by oxygen-starving mitochondria inhibit hydroxylation and HIFαs escape degradation, accumulate, and translocate inside the nucleus where they assemble with ARNT into transcriptionally active heterodimers (Figure 1) [26].
3. Oxygen-Independent Regulation of HIFs
Over the past few years, it has become clear that HIFs can also be regulated by mechanisms not directly affected by oxygen concentration. This regulation can occur at multiple levels including transcription, translation, post-translational modification, stabilization, nuclear translocation and activation of HIFαs [27][26][28][29]. Post-translational modifications of HIFαs probably hold the most important role in their regulation [30]. HIFαs are subjected to acetylation, s-nitrosylation and sumoylation, but the significance of these modifications for HIF activity is still a matter of debate. On the other hand, HIFα phosphorylation is much better characterized with clearly demonstrated importance in various cellular models. Both HIF-1α and HIF-2α can be directly phosphorylated by several kinases including GSK3, PLK3, ATM, PKA, CDK1, and the extent of their modification depends on cell type and extracellular signals [30][31]. Previous work from our lab has also shown that HIF-1α is a direct target of kinases ERK1/2 and CK1δ, modification by which has distinct outcomes on HIF-1 activity [32][33][34][35]. More specifically, while import of HIF-1α inside the nucleus is constitutive, mediated by the importin α/β as well as importins 4/7 [36][37], nuclear export of HIF-1α is regulated in an ERK1/2- and CRM1-dependent manner (Figure 1) [34][35]. Phosphorylation of HIF-1α by ERK1/2 at Ser641/643, which lay inside a small domain termed ETD (ERK Targeted Domain; amino acids 616–658), masks a nearby CRM1-dependent nuclear export signal (NES), thus inhibiting HIF-1α nuclear export and increasing HIF-1α nuclear concentration and HIF-1 transcriptional activity [34][35]. Lack of this phosphorylation allows CRM1 binding to HIF-1α and its subsequent translocation to the cytoplasm, where, interestingly, HIF-1α interacts with mortalin and takes part in the assembly of anti-apoptotic complex on the surface of the mitochondria [32][38]. This mode of HIF-1α regulation by ERK1/2 has been exploited for the development of peptide HIF-1 inhibitors modelled after the ETD amino acid sequence. On the other hand, phosphorylation of HIF-1α by Ck1δ at Ser247 inside the PAS domain has a negative effect by inhibiting the ability of HIF-1α to associate with ARNT [33][34][35][39][40][41]. Regulation of the HIF-1 heterodimer assembly by phosphorylation as well as by MgcRacGAP [42][43] highlights the HIF-1α/ARNT interaction as a target for peptide inhibitors modelled after the PAS domain, an approach that has indeed been successfully tried (see also below). ERK1/2 and CK1δ also modify HIF-2α at distinct sites, and in this case, they both appear to regulate HIF-2 activity by affecting the distribution of HIF-2α between nucleus and cytoplasm [40][41].
In addition to direct phosphorylation, signaling pathways involving PI3K, ERK1/2 or p38 MAPK when activated by non-hypoxic stimuli such as growth factors (e.g., PDGF, TGF-β, IGF-1, and EGF), cytokines or hormones can also affect HIF activation by indirectly modulating its expression or stability [30][31][44][45][46][47][48][49][50]. As an example, heregulin (a member of the EGF family of growth factors) induces HIF-1 by activating the PI3K/Akt/mTOR pathway and increasing the rate of HIF-1α translation [51]. Other exemplary modes of HIF regulation include the involvement of ROS signaling (reviewed in [52]), transcription factors such as NF-kb [53] and STAT3 [54] that upregulate transcription of the gene encoding HIF-1α and many interacting proteins such as HSP90 and RACK1, which stabilize or destabilize HIF-1α, respectively [55][56][57]. HIFα stability is also affected by CO2 concentration as both in vivo and in vitro hypercapnia decrease HIFα protein levels independently of the PHD/pVHL-mediated degradation pathway and, most likely, via lysosomal proteolysis [58].
4. The Involvement of HIFs in Cancer
HIFs and especially HIF-1 influence several hallmarks of cancer such as genomic instability, tumor cell invasion, metastasis, and angiogenesis as well as suppression of the anti-tumor immune response [59][60]. Most importantly, HIF-1 holds a prominent role as mediator of the metabolic reprogramming that characterizes many types of cancer cells [1][61]. HIF-1 upregulates expression of most enzymes of glycolysis as well as expression of pyruvate dehydrogenase kinase 1 (PDK1). PDK1 phosphorylates and inactivates pyruvate dehydrogenase (PDH), which catalyzes conversion of pyruvate into acetyl-CoA. Thus, HIF-1 drives pyruvate, the product of glycolysis away from the TCA cycle and towards production of lactate, even in the presence of oxygen, a phenomenon known as Warburg effect [1][61]. Lipid metabolism is also influenced by a HIF-1 and several HIF-1 gene targets are involved in lipogenesis, which is generally favored in cancer via an increase in fatty acid uptake or synthesis and storage and simultaneous downregulation of fatty acid oxidation (reviewed in [62]). This often leads to accumulation of lipid droplets [33][63][64][65] and protects cancer cells from lipotoxicity [33][63][64]. HIF-dependent gene expression is also important for the adaptation and metabolism of cells surrounding a tumor (e.g., stromal cells), which are known to play an important role for cancer development [61][66].
There are numerous studies in which HIF-α proteins are found overexpressed in malignant tumors [59]. In principle, overexpression of HIFα isoforms is associated with poor clinical outcomes in patients with solid tumors [59][15]. Interestingly, HIF-1α was also found elevated and correlated with bad prognosis in hematological malignancies (reviewed in [67]). However, there are few reports indicating that HIF-1α overexpression may be connected to a positive outcome in certain cancer types including head and neck [68], non-small cell lung [69] and neuroblastoma [70].
Another feature that gives HIFs a special role in cancer progression is their ability to promote epithelial to mesenchymal transition (EMT) as well as resistance to chemo- or radio-therapy. HIFs facilitate EMT mainly by enhancing the expression of genes such as TCF3, ZFHX1A/B, and TWIST, which repress E-cadherin and epithelial type promoting factors, while, at the same time, the expression of mesenchymal type genes is increased [71][72]. Furthermore, HIF-mediated gene expression drives extracellular matrix remodeling, resistance to anoikis-related cell death and establishment of new cancer colonies, all of which facilitate the metastatic phenotype of hypoxic tumors [73]. Moreover, HIF-1 mediates chemoresistance by inducing expression of proteins that enhance drug efflux such as multidrug resistance 1 (MDR1) [74][75] and MRP2 [76] or anti-apoptotic proteins that promote drug resistance such as survivin [77][78]. HIF-1 is also implicated in resistance to radiation therapy since it counteracts the cytotoxic effects of radiation such as DNA damage and production of reactive oxygen species (ROS) [79][80].
Despite the unequivocal involvement of HIFs in the adaptation of cancer cells in the hypoxic tumor microenvironment, which promotes tumor progression, metastasis and resistance to therapy, it is still questionable whether HIFs by themselves are pro-oncogenic in a normal genetic background [81]. HIFs are active under physiological conditions such as embryonic development, immune system development, high-altitude adaptation and exercise and play an essential role in the maintenance of normal tissue homeostasis. HIF activation under these conditions does not trigger oncogenesis. Even when HIFα is constitutively activated due to pVHL function loss in renal cells, development of renal carcinoma requires additional mutations [82]. These issues have become especially important due the recent licensing and wide clinical administration of PHD inhibitors as HIF activators, and subsequent erythropoiesis inducers, for the treatment of patients suffering from renal anemia [83]. Long-term administration of these PHD inhibitors has not demonstrated tumor-initiating or tumor-promoting effects in either animal models or phase III clinical trials, possibly because competitive inhibition of PHD catalytic activity cannot cause permanent and irreversible HIF activation or pharmacological HIF induction is graded and cannot exceed a physiologically acceptable threshold. Nevertheless, the proof-of-principle for HIF inhibition as a valid anticancer strategy has been demonstrated in several cases and in both animal and human studies.
References
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15.
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 1992, 12, 5447–5454.
- Wang, G.L.; Semenza, G.L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. USA 1993, 90, 4304–4308.
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237.
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82.
- Skuli, N.; Majmundar, A.J.; Krock, B.L.; Mesquita, R.C.; Mathew, L.K.; Quinn, Z.L.; Runge, A.; Liu, L.; Kim, M.N.; Liang, J.; et al. Endothelial HIF-2alpha regulates murine pathological angiogenesis and revascularization processes. J. Clin. Investig. 2012, 122, 1427–1443.
- Heikkila, M.; Pasanen, A.; Kivirikko, K.I.; Myllyharju, J. Roles of the human hypoxia-inducible factor (HIF)-3alpha variants in the hypoxia response. Cell Mol. Life Sci. 2011, 68, 3885–3901.
- Makino, Y.; Cao, R.; Svensson, K.; Bertilsson, G.; Asman, M.; Tanaka, H.; Cao, Y.; Berkenstam, A.; Poellinger, L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 2001, 414, 550–554.
- Tolonen, J.P.; Heikkila, M.; Malinen, M.; Lee, H.M.; Palvimo, J.J.; Wei, G.H.; Myllyharju, J. A long hypoxia-inducible factor 3 isoform 2 is a transcription activator that regulates erythropoietin. Cell Mol. Life Sci. 2020, 77, 3627–3642.
- Pugh, C.W.; Ratcliffe, P.J. New horizons in hypoxia signaling pathways. Exp. Cell Res. 2017, 356, 116–121.
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514.
- Wu, D.; Potluri, N.; Lu, J.; Kim, Y.; Rastinejad, F. Structural integration in hypoxia-inducible factors. Nature 2015, 524, 303–308.
- Key, J.; Scheuermann, T.H.; Anderson, P.C.; Daggett, V.; Gardner, K.H. Principles of ligand binding within a completely buried cavity in HIF2alpha PAS-B. J. Am. Chem. Soc. 2009, 131, 17647–17654.
- Lando, D.; Gorman, J.J.; Whitelaw, M.L.; Peet, D.J. Oxygen-dependent regulation of hypoxia-inducible factors by prolyl and asparaginyl hydroxylation. Eur. J. Biochem. 2003, 270, 781–790.
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 2019, 14, 667–682.
- Hu, C.-J.; Sataur, A.; Wang, L.; Chen, A.; Simon, M.C. The N-Terminal Transactivation Domain Confers Target Gene Specificity of Hypoxia-inducible Factors HIF-1α and HIF-2α. Mol. Biol. Cell 2007, 18, 4528–4552.
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275.
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472.
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468.
- Wilson, J.W.; Shakir, D.; Batie, M.; Frost, M.; Rocha, S. Oxygen-sensing mechanisms in cells. FEBS J. 2020.
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54.
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340.
- Berra, E.; Benizri, E.; Ginouves, A.; Volmat, V.; Roux, D.; Pouyssegur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003, 22, 4082–4090.
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686.
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471.
- Schödel, J.; Ratcliffe, P.J. Mechanisms of hypoxia signalling: New implications for nephrology. Nat. Rev. Nephrol. 2019, 15, 641–659.
- Semenza, G.L. The Genomics and Genetics of Oxygen Homeostasis. Annu Rev. Genom. Hum. Genet. 2020, 21, 183–204.
- Ivanova, I.G.; Park, C.V.; Kenneth, N.S. Translating the Hypoxic Response-the Role of HIF Protein Translation in the Cellular Response to Low Oxygen. Cells 2019, 8, 114.
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389.
- Albanese, A.; Daly, L.A.; Mennerich, D.; Kietzmann, T.; See, V. The Role of Hypoxia-Inducible Factor Post-Translational Modifications in Regulating Its Localisation, Stability, and Activity. Int. J. Mol. Sci. 2020, 22, 268.
- Kietzmann, T.; Mennerich, D.; Dimova, E.Y. Hypoxia-Inducible Factors (HIFs) and Phosphorylation: Impact on Stability, Localization, and Transactivity. Front. Cell Dev. Biol 2016, 4, 11.
- Karagiota, A.; Kourti, M.; Simos, G.; Mylonis, I. HIF-1alpha-derived cell-penetrating peptides inhibit ERK-dependent activation of HIF-1 and trigger apoptosis of cancer cells under hypoxia. Cell Mol. Life Sci. 2019, 76, 809–825.
- Kourti, M.; Ikonomou, G.; Giakoumakis, N.N.; Rapsomaniki, M.A.; Landegren, U.; Siniossoglou, S.; Lygerou, Z.; Simos, G.; Mylonis, I. CK1delta restrains lipin-1 induction, lipid droplet formation and cell proliferation under hypoxia by reducing HIF-1alpha/ARNT complex formation. Cell Signal. 2015, 27, 1129–1140.
- Mylonis, I.; Chachami, G.; Paraskeva, E.; Simos, G. Atypical CRM1-dependent nuclear export signal mediates regulation of hypoxia-inducible factor-1alpha by MAPK. J. Biol. Chem. 2008, 283, 27620–27627.
- Mylonis, I.; Chachami, G.; Samiotaki, M.; Panayotou, G.; Paraskeva, E.; Kalousi, A.; Georgatsou, E.; Bonanou, S.; Simos, G. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J. Biol. Chem. 2006, 281, 33095–33106.
- Chachami, G.; Paraskeva, E.; Mingot, J.M.; Braliou, G.G.; Gorlich, D.; Simos, G. Transport of hypoxia-inducible factor HIF-1alpha into the nucleus involves importins 4 and 7. Biochem. Biophys. Res. Commun. 2009, 390, 235–240.
- Depping, R.; Steinhoff, A.; Schindler, S.G.; Friedrich, B.; Fagerlund, R.; Metzen, E.; Hartmann, E.; Kohler, M. Nuclear translocation of hypoxia-inducible factors (HIFs): Involvement of the classical importin alpha/beta pathway. Biochim. Biophys. Acta 2008, 1783, 394–404.
- Mylonis, I.; Kourti, M.; Samiotaki, M.; Panayotou, G.; Simos, G. Mortalin-mediated and ERK-controlled targeting of HIF-1alpha to mitochondria confers resistance to apoptosis under hypoxia. J. Cell Sci. 2017, 130, 466–479.
- Kalousi, A.; Mylonis, I.; Politou, A.S.; Chachami, G.; Paraskeva, E.; Simos, G. Casein kinase 1 regulates human hypoxia-inducible factor HIF-1. J. Cell Sci. 2010, 123, 2976–2986.
- Gkotinakou, I.M.; Befani, C.; Simos, G.; Liakos, P. ERK1/2 phosphorylates HIF-2alpha and regulates its activity by controlling its CRM1-dependent nuclear shuttling. J. Cell Sci. 2019, 132.
- Pangou, E.; Befani, C.; Mylonis, I.; Samiotaki, M.; Panayotou, G.; Simos, G.; Liakos, P. HIF-2alpha phosphorylation by CK1delta promotes erythropoietin secretion in liver cancer cells under hypoxia. J. Cell Sci. 2016, 129, 4213–4226.
- Lyberopoulou, A.; Mylonis, I.; Papachristos, G.; Sagris, D.; Kalousi, A.; Befani, C.; Liakos, P.; Simos, G.; Georgatsou, E. MgcRacGAP, a cytoskeleton regulator, inhibits HIF-1 transcriptional activity by blocking its dimerization. Biochim. Biophys. Acta 2013, 1833, 1378–1387.
- Lyberopoulou, A.; Venieris, E.; Mylonis, I.; Chachami, G.; Pappas, I.; Simos, G.; Bonanou, S.; Georgatsou, E. MgcRacGAP interacts with HIF-1alpha and regulates its transcriptional activity. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2007, 20, 995–1006.
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545.
- Stiehl, D.P.; Jelkmann, W.; Wenger, R.H.; Hellwig-Burgel, T. Normoxic induction of the hypoxia-inducible factor 1alpha by insulin and interleukin-1beta involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002, 512, 157–162.
- Beppu, K.; Nakamura, K.; Linehan, W.M.; Rapisarda, A.; Thiele, C.J. Topotecan blocks hypoxia-inducible factor-1alpha and vascular endothelial growth factor expression induced by insulin-like growth factor-I in neuroblastoma cells. Cancer Res. 2005, 65, 4775–4781.
- Calvani, M.; Trisciuoglio, D.; Bergamaschi, C.; Shoemaker, R.H.; Melillo, G. Differential involvement of vascular endothelial growth factor in the survival of hypoxic colon cancer cells. Cancer Res. 2008, 68, 285–291.
- Mohlin, S.; Hamidian, A.; von Stedingk, K.; Bridges, E.; Wigerup, C.; Bexell, D.; Pahlman, S. PI3K-mTORC2 but not PI3K-mTORC1 regulates transcription of HIF2A/EPAS1 and vascularization in neuroblastoma. Cancer Res. 2015, 75, 4617–4628.
- Gorlach, A.; Diebold, I.; Schini-Kerth, V.B.; Berchner-Pfannschmidt, U.; Roth, U.; Brandes, R.P.; Kietzmann, T.; Busse, R. Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: Role of the p22(phox)-containing NADPH oxidase. Circ. Res. 2001, 89, 47–54.
- Richard, D.E.; Berra, E.; Pouyssegur, J. Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1alpha in vascular smooth muscle cells. J. Biol. Chem. 2000, 275, 26765–26771.
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: Novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell Biol. 2001, 21, 3995–4004.
- Gorlach, A.; Kietzmann, T. Superoxide and derived reactive oxygen species in the regulation of hypoxia-inducible factors. Methods Enzym. 2007, 435, 421–446.
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811.
- Papadakis, A.I.; Paraskeva, E.; Peidis, P.; Muaddi, H.; Li, S.; Raptis, L.; Pantopoulos, K.; Simos, G.; Koromilas, A.E. eIF2alpha Kinase PKR modulates the hypoxic response by Stat3-dependent transcriptional suppression of HIF-1alpha. Cancer Res. 2010, 70, 7820–7829.
- Amir, S.; Wang, R.; Simons, J.W.; Mabjeesh, N.J. SEPT9_v1 up-regulates hypoxia-inducible factor 1 by preventing its RACK1-mediated degradation. J. Biol. Chem. 2009, 284, 11142–11151.
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol. Cell 2007, 25, 207–217.
- Semenza, G.L. A compendium of proteins that interact with HIF-1alpha. Exp. Cell Res. 2017, 356, 128–135.
- Selfridge, A.C.; Cavadas, M.A.; Scholz, C.C.; Campbell, E.L.; Welch, L.C.; Lecuona, E.; Colgan, S.P.; Barrett, K.E.; Sporn, P.H.; Sznajder, J.I.; et al. Hypercapnia Suppresses the HIF-dependent Adaptive Response to Hypoxia. J. Biol. Chem. 2016, 291, 11800–11808.
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770.
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790.
- Nakazawa, M.S.; Keith, B.; Simon, M.C. Oxygen availability and metabolic adaptations. Nat. Rev. Cancer 2016, 16, 663–673.
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214.
- Mylonis, I.; Sembongi, H.; Befani, C.; Liakos, P.; Siniossoglou, S.; Simos, G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J. Cell Sci. 2012, 125, 3485–3493.
- Yoo, W.; Noh, K.H.; Ahn, J.H.; Yu, J.H.; Seo, J.A.; Kim, S.G.; Choi, K.M.; Baik, S.H.; Choi, D.S.; Kim, T.W.; et al. HIF-1alpha expression as a protective strategy of HepG2 cells against fatty acid-induced toxicity. J. Cell. Biochem. 2014, 115, 1147–1158.
- Triantafyllou, E.A.; Georgatsou, E.; Mylonis, I.; Simos, G.; Paraskeva, E. Expression of AGPAT2, an enzyme involved in the glycerophospholipid/triacylglycerol biosynthesis pathway, is directly regulated by HIF-1 and promotes survival and etoposide resistance of cancer cells under hypoxia. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1142–1152.
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593.
- Deynoux, M.; Sunter, N.; Herault, O.; Mazurier, F. Hypoxia and Hypoxia-Inducible Factors in Leukemias. Front. Oncol. 2016, 6, 41.
- Beasley, N.J.; Leek, R.; Alam, M.; Turley, H.; Cox, G.J.; Gatter, K.; Millard, P.; Fuggle, S.; Harris, A.L. Hypoxia-inducible factors HIF-1alpha and HIF-2alpha in head and neck cancer: Relationship to tumor biology and treatment outcome in surgically resected patients. Cancer Res. 2002, 62, 2493–2497.
- Volm, M.; Koomagi, R. Hypoxia-inducible factor (HIF-1) and its relationship to apoptosis and proliferation in lung cancer. Anticancer Res. 2000, 20, 1527–1533.
- Noguera, R.; Fredlund, E.; Piqueras, M.; Pietras, A.; Beckman, S.; Navarro, S.; Pahlman, S. HIF-1alpha and HIF-2alpha are differentially regulated in vivo in neuroblastoma: High HIF-1alpha correlates negatively to advanced clinical stage and tumor vascularization. Clin. Cancer Res. 2009, 15, 7130–7136.
- Krishnamachary, B.; Zagzag, D.; Nagasawa, H.; Rainey, K.; Okuyama, H.; Baek, J.H.; Semenza, G.L. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006, 66, 2725–2731.
- Yang, M.-H.; Wu, M.-Z.; Chiou, S.-H.; Chen, P.-M.; Chang, S.-Y.; Liu, C.-J.; Teng, S.-C.; Wu, K.-J. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305.
- Schito, L.; Rey, S. Hypoxic pathobiology of breast cancer metastasis. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 239–245.
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394.
- Ding, Z.; Yang, L.; Xie, X.; Xie, F.; Pan, F.; Li, J.; He, J.; Liang, H. Expression and significance of hypoxia-inducible factor-1 alpha and MDR1/P-glycoprotein in human colon carcinoma tissue and cells. J. Cancer Res. Clin. Oncol. 2010, 136, 1697–1707.
- Krishnamurthy, P.; Schuetz, J.D. The ABC transporter Abcg2/Bcrp: Role in hypoxia mediated survival. Biometals 2005, 18, 349–358.
- Chen, Y.Q.; Zhao, C.L.; Li, W. Effect of hypoxia-inducible factor-1alpha on transcription of survivin in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2009, 28, 29.
- Faversani, A.; Vaira, V.; Moro, G.P.; Tosi, D.; Lopergolo, A.; Schultz, D.C.; Rivadeneira, D.; Altieri, D.C.; Bosari, S. Survivin family proteins as novel molecular determinants of doxorubicin resistance in organotypic human breast tumors. Breast Cancer Res. 2014, 16, R55.
- Befani, C.; Liakos, P. The role of hypoxia-inducible factor-2 alpha in angiogenesis. J. Cell Physiol. 2018.
- Karakashev, S.V.; Reginato, M.J. Progress toward overcoming hypoxia-induced resistance to solid tumor therapy. Cancer Manag. Res. 2015, 7, 253–264.
- Cummins, E.P.; Strowitzki, M.J.; Taylor, C.T. Mechanisms and Consequences of Oxygen and Carbon Dioxide Sensing in Mammals. Physiol. Rev. 2020, 100, 463–488.
- Jonasch, E.; Walker, C.L.; Rathmell, W.K. Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat. Rev. Nephrol. 2020.
- Sanghani, N.S.; Haase, V.H. Hypoxia-Inducible Factor Activators in Renal Anemia: Current Clinical Experience. Adv. Chronic Kidney Dis. 2019, 26, 253–266.


