 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Umesh Kalathiya | + 2280 word(s) | 2280 | 2021-03-09 06:54:52 | | | |
| 2 | Bruce Ren | Meta information modification | 2280 | 2021-03-16 02:26:55 | | |
Video Upload Options
The fundamentals of how protein–protein/RNA/DNA interactions influence the structures and functions of the workhorses from the cells have been well documented in the 20th century. A diverse set of methods exist to determine such interactions between different components, particularly, the mass spectrometry (MS) methods, with its advanced instrumentation, has become a significant approach to analyze a diverse range of biomolecules, as well as bring insights to their biomolecular processes. Cross-linking mass spectrometry (CLMS) holds promise to identify interaction sites in larger and more complex biological systems.
1. Introduction
Decades of research into the cell biology, molecular biology, biochemistry, structural biology, and biophysics have produced a detailed understanding of individual DNA/RNA/protein molecules, and their interconnected networks. A great diversity of techniques has emerged for studying their structural interactions. However, even more complex, the structures of these workhorses from the cells are themselves dynamic, converting from one dominant form to another based on the proportions of particular proteoforms present for any given biomolecules. Accordingly, beyond the protein–protein/DNA/RNA interaction landscape, there is an entire universe to explore with respect to their structure and dynamics. One such high-throughput technique has emerged as a dominant player in understanding both interaction landscapes and their resulting protein/DNA/RNA structures, namely cross-linking mass spectrometry (CLMS). This review covers different methods that are available to study protein–protein/DNA/RNA interactions, and provides vital insight into CLMS, a collection of methods that are perfectly suited to achieve a better understanding of intra- or inter- molecular interactions.
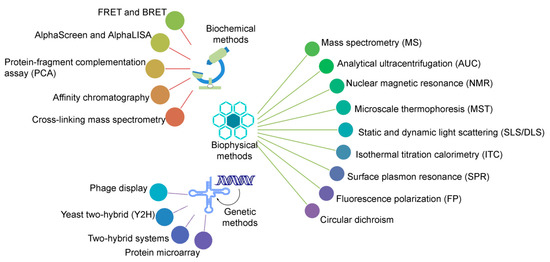
Protein–protein interactions (PPIs) play a crucial role in all biological/biomolecular processes to understand the molecular mechanism of relevant protein molecules; hence, they are often termed the workhorses of cells. Over 80% of proteins do not function in isolation, but rather exist in interactions with one another to obtain stable or transitory complexes, as demanded by their observed function [1][2]. PPIs are considered to be an emerging class of drug target, since aberrant protein–protein interactions can participate in the pathogenesis of various human diseases, which, in turn, can contribute significant options for diagnostic as well as therapeutic targets. A large number of experimental methods have been developed to study PPIs, based on biophysical, biochemical, or genetic principles (as shown in Figure 1); and, each individual type has advantages as well as limitations regarding structural coverage of amino acid sequences, sensitivity, and specificity [1][3][4][5][6]. Methods can be chosen to emphasize different aspects of PPIs, such as identifying a protein binding partner(s), generating structural details of protein complexes, analyzing kinetic and thermodynamic constants of interactions, visualizing and quantifying PPIs in real time in living cells, and mapping small interactomes that refer to specific cellular pathways [1][3][4][5].
Mass spectrometry (MS) is one of the powerful approaches available that is useful in several areas beyond CLMS (e.g., hydrogen-deuterium exchange [7]), and it is particularly useful in combination with other techniques, providing steady progress for structural biology. Technical advances in mass spectrometry have made it possible to study protein–protein interactions from simple protein complexes to wide scale proteome experiments, which were not earlier accessible by traditional techniques [8][9][10]. Above all, mass spectrometry methods have democratized protein interaction analysis by making them accessible, relatively inexpensive, and high throughput. Having these advantages MS is becoming progressively popular in the structural biology stream for analyzing three dimensional (3D) structures and mapping their interactions with partner molecules. While emerging techniques, like cryoEM, can summarize large numbers of protein complexes, much of the resulting dynamics of the structure is missed and such gaps in structural data sets can be bridged by low-resolution methods, for example, chemical cross-linking (CL) [11][12][13]. The overall architecture of a protein complex can be obtained through electron microscopy (EM) [14], small-angle X-ray scattering (SAXS) [15], and ion-mobility (IM) MS [16], whereas the precise residues forming protein–protein interactions can be identified using hydrogen-deuterium exchange [17], chemical cross-linking [11][12][13], and chemical foot printing [18].
2. Function of CLMS
Fundamentally, the mass spectrometry methods simply measure a mass-to-charge ratio of an ion. Initially, analytes are ionized and then transferred into the gas phase prior to their separation according to mass-to-charge ratios in a mass analyzer. Subsequently, ions that emerge from the mass analyzer are recorded using a detector. The most common way to ionize a peptide or protein sample is electrospray ionization (ESI) [41]. However, a complementary ionization technique, known as matrix-assisted laser desorption/ionization (MALDI), is often used for its relative ease of use for novices. In a few studies analyzing intact protein complexes, MALDI mass spectrometry (MALDI-MS) has been used together with chemical cross-linking techniques [42][43][44]. Regardless of ionization methods, there are various types of mass analyzers accessible, including the following common ones: ion trap, time-of-flight (TOF), quadrupole, and orbitrap. Each of these mass analyzers may be configured in various ways with similar or different mass analyzers to form unique types of mass spectrometers [45].
Quantitative cross-linking mass spectrometry (QCLMS) approaches investigate protein structures as well as the dynamics of their interactions [46][47][48][49][50][51]. QCLMS is often performed using a unique cross-linker that introduces a corresponding mass shift after isotope labeling specific only to the cross-linked peptides [46][47][50][52][53], followed by quantitation of cross-links in MS1. However, the limited availability of isotope labeled cross-linkers restrains the implementation of this approach in QCLMS [54]. Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC), which is a popular option is an alternative to the QCLMS approach, which relies on the metabolic incorporation of isotope labeled amino acids from culture media. Metabolically labeled samples are cross-linked and pooled; relative quantitation is performed from MS1 data using the characteristic mass shift introduced into peptides from the incorporation of isotope labeled amino acid/s [48]. SILAC enables a comparison of multiple samples per analysis (usually two) and it can also enable monitoring amino acid incorporation in a time course (pulsed SILAC), which is especially valuable for exploring dynamics in biological processes. Recently, iCLASPI (in vivo cross-linking assisted and stable isotope labeling by amino acids in cell culture (SILAC)-based protein identification), an approach combining SILAC and in vivo cross-linking, has been implemented to quantify native protein–protein interactions in HEK293T cells. iCLASPI has been successfully implemented to profile native protein–protein interactions involving core histones H3 and H4 in biological context [55]. Chavez et al. successfully implemented SILAC and cross-linking to investigate key protein–protein interactions, and then investigated the Hsp90 conformational changes upon treatment with 17-AAG Hsp90 N-terminal domain inhibitor [49].
Isobaric Tags for Relative and Absolute Quantitation/Tandem Mass Tag (iTRAQ/TMT) are chemical labeling, which are introduced to peptides after protease digestion, which allow relative protein quantitation via MS2 or MS3 encoded data. Nowadays, TMT/iTRAQ labeling enables comparison of up to 16 samples (valid for TMT) in a single MS analysis. Notably, the iTRAQ/TMT reporter ions used for quantitation are cleaved from labeled peptides during fragmentation by way of collision-induced dissociation allowing for the quantitation from fragment ion relative intensities. Yu et al. [56], implemented the TMT approach in a multiplexed comparison of protein complex dynamics and protein–protein interactions. Their QMIX (Quantitation of Multiplexed, Isobaric-labeled cross (X)-linked peptides) workflow with TMT labeling, achieves peptide quantitation from MS3 data that eliminates interference from ions that were observed in MS1 data along with isotope labeled cross-linkers or SILAC cross-linking [56]. Furthermore, the precise MS2 quantitation of protein cross-linking could also be achieved in a label-free manner utilizing a data-independent acquisition mode (DIA). The extraction of cross-linked peptide quantities from DIA data is usually performed utilizing a spectral library prepared particularly from investigated samples. Muller et al. developed a novel DIA-QCLMS approach utilizing photo activatable cross-linkers ensuring reliable quantitation of cross-linked proteins across a wide range of environmental changes, such as pH, temperature pressure, or concentration [51][52][57]. DIA approaches promise to be useful in future applications of quantitative cross-linking proteomics, due to their precision, reproducibility, and label-free manner.
Moreover, techniques, such as proximity-dependent biotin labeling (or BioID technique) in living cells, help to understand the plasticity of protein networks within heterogeneous cellular populations. In combination with nanopore technology, such an approach could help tackle pending biological questions, e.g., the identification of peptidyl-prolyl isomerases (PPIases) substrates. PPIases substrates preserve their primary structure/molecular mass, as well as the cis and trans isomers of the proline peptide bonds of the substrates interconversion by PPIases being the sole change. This subtle modification triggers important changes to the substrate’s fate, such as subcellular translocation, degradation, or rewiring of their protein–protein interaction networks. PPIase enzymes act as central molecular switches, as exemplified by the peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin1) that has been extensively studied and showing its involvement in multiple diseases [58][59][60]. Herein, we propose that the CLMS techniques, though, which have not been previously carried out on such problems, could be merged with novel interactomics techniques (proximity-dependent labeling by BioID or an engineered biotin ligase by TurboID technique). Combining these two approaches may bring spatial resolution to CLMS at a sub organelle level, since the BioID radius is estimated ~10 nm and CLMS is aimed on the proteins in the neighborhood of a given bait.
While there has been some success in the proteome wide CLMS [61][62][63][64], in the case of both chemical cross-linking MS and the related techniques of native MS [65][66], a prior purification of single protein or protein complex is typically necessary, and this can be acquired through over expression with the purification of a recombinant version of particular system. When investigating the protein assemblies, individual components need to be purified to reconstitute the whole complex later in vitro and, alternatively, in vivo reconstitution can occur by co-expressing various subunits [67]. Because the reconstituted systems mostly benefit from large yields to aid the structural analysis and they are frequently done using bacteria (mostly E. coli) as the host system, functionally important post-translational modifications and interacting protein partner associations may be lost during this process. Thus, various biochemical approaches must be explored when beginning a new project in order to directly enable the isolation of endogenous protein complexes from cells or tissues [68].
Modern mass spectrometry that is coupled with the chemical cross-linking of juxtaposed amino acids can provide important structural information. Two main types of the cross-linking strategies involving either the activation of the cross-linking reagent by UV or chemical methods to enable cross-linking [11][12][13][69]. Chemical cross-linking is a classical approach for determining protein–protein interactions and is also one of the first approaches that has been used to map large complexes, for example, the ribosome [2]. Generally, the cross-linking techniques link two or more proteins present in a complex by covalent bonds and, as the name implies, via a molecule designed to bridge juxtaposed amino acids, i.e., to chemically cross-link residues. The chosen cross-linker is a chemical reagent that contains two or more reactive groups connected through a spacer or linker of various lengths [70]. By using this method, low-affinity protein–protein contacts, or some specific interactions, can be detected that are difficult to characterize by other methods (e.g., nuclear magnetic resonance (NMR), X-Ray, etc.). Moreover, the cross-linking techniques have also been applied to stabilize transient protein–protein interactions in a dynamic process both in vitro and in vivo [1][2]. However, there can be considerable weakness in these chemical methods that are related to the lack of spatial localization in a cell and a lack of control over activity. Thus, to evaluate PPIs as similarly as possible to the native conditions, photo-cross linking methods are valuable due to their ability to generate reactive species in situ instantaneously by irradiation with UV-light [11][12][13][69].
Cross-linking is always followed by other downstream methods to further analyze the cross-linked proteins, often using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) to separate the cross-linked from non-cross-linked proteins, tandem affinity purification (TAP) or immunoaffinity chromatography for affinity-based purification of cross-linked products, and mass spectrometry methods for the interacting partner identification [71][72]. Additionally, SDS-PAGE analysis is very helpful in the early stages of analysis when empirically working out the correct ratio of cross-linker to protein complex. A limitation of using chemical cross-linking while using these methods is the high risk of detecting non-specific interactions. However, these limitations can be addressed using more than one cross-linker of differing activities or spanning various distances and by varying the ratio of reagent to protein complex. Non-specific interactions can result from proteins in close proximity that may not be functionally related. These suggest that while the CLMS technique is relatively straightforward to implement, the identification of relevant cross-linked proteins could be quite demanding due to the intracellular dynamic range of expression of proteins, which can range from one to one million copies low abundance of cross-linked species [1].
Innovative developments in the biological applications of MS led to the development of a large number of methods, and it has made it relatively simple to identify proteins alone or in complexes using CLMS technique. Large macromolecular complexes like ribosomes or exosomes, have been purified and analyzed directly using mass spectrometry [73][74][75][76]. Recently, chemical cross-linking combined with mass spectrometry based structural techniques have hit their stride allowing for various biologically relevant molecular machines to be successfully studied in the past few years using this combination [77]. The various workflows that have been developed to implement CLMS represent a vast toolkit that can help to provide novel insight into the structure and organization of proteins in order to define protein–protein interactions and probing PPI interfaces.
References
- Zhou, M.; Li, Q.; Wang, R. Current experimental methods for characterizing protein-protein interactions. Chem. Med. Chem. 2016, 11, 738–756.
- Berggård, T.; Linse, S.; James, P. Methods for the detection and analysis of protein-protein interactions. Proteomics 2007, 16, 2833–2842.
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 12, 862–873.
- Joerger, A.C.; Fersht, A.R. Structure-function-rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242.
- Brooke, M.A.; Nitoiu, D.; Kelsell, D.P. Cell-cell connectivity: Desmosomes and disease. J. Pathol. 2012, 226, 158–171.
- Heck, A.J. Native mass spectrometry: A bridge between interactomics and structural biology. Nat. Methods 2008, 5, 927–933.
- Pirrone, G.F.; Iacob, R.E.; Engen, J.R. Applications of hydrogen/deuterium exchange MS from 2012 to 2014. Anal. Chem. 2015, 87, 99–118.
- Tugarinov, V.; Hwang, P.M.; Kay, L.E. Nuclear magnetic resonance spectroscopy of high-molecular-weight proteins. Annu. Rev. Biochem. 2004, 73, 107–146.
- Ilari, A.; Savino, C. Protein structure determination by x-ray crystallography. Methods Mol. Biol. 2008, 452, 63–87.
- Walian, P.; Cross, T.A.; Jap, B.K. Structural genomics of membrane proteins. Genome Biol. 2004, 5, 215.
- Iacobucci, C.; Götze, M.; Sinz, A. Cross-linking/mass spectrometry to get a closer view on protein interaction networks. Curr. Opin. Biotechnol. 2020, 63, 48–53.
- Hino, N.; Okazaki, Y.; Kobayashi, T.; Hayashi, A.; Sakamoto, K.; Yokoyama, S. Protein photo-cross-linking in mammalian cells by site-specific incorporation of a photoreactive amino acid. Nat. Methods 2005, 2, 201–206.
- Farrell, I.S.; Toroney, R.; Hazen, J.L.; Mehl, R.A.; Chin, J.W. Photo-cross-linking interacting proteins with a genetically encoded benzophenone. Nat. Methods 2005, 2, 377–384.
- Milne, J.L.; Borgnia, M.J.; Bartesaghi, A.; Tran, E.E.; Earl, L.A.; Schauder, D.M.; Lengyel, J.; Pierson, J.; Patwardhan, A.; Subramaniam, S. Cryo-electron microscopy-a primer for the nonmicroscopist. FEBS J. 2013, 280, 28–45.
- Hura, G.L.; Menon, A.L.; Hammel, M.; Rambo, R.P.; Poole, F.L., II; Tsutakawa, S.E.; Jenney, F.E., Jr.; Classen, S.; Frankel, K.A.; Hopkins, R.C.; et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nat. Methods 2009, 6, 606–612.
- Uetrecht, C.; Rose, R.J.; Van Duijn, E.; Lorenzen, K.; Heck, A.J. Ion mobility mass spectrometry of proteins and protein assemblies. Chem Soc. Rev. 2010, 39, 1633–1655.
- Konermann, L.; Pan, J.; Liu, Y.H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 2011, 40, 1224–1234.
- Gau, B.C.; Sharp, J.S.; Rempel, D.L.; Gross, M.L. Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal. Chem. 2009, 81, 6563–6571.
- Perrin, F. Polarisation of fluorescence and mean life of excited molecules. Polarisation de la lumière de fluorescence. Vie moyenne des molécules dans l’etat excite. J. Phys. Radium. 1926, 7, 390–401.
- Du, Y. Protein-Protein Interactions, of the Series Methods in Molecular Biology; Meyerkord, C.L., Fu, H., Walker, J.M., Eds.; Springer: New York, NY, USA, 2015; Volume 1278, pp. 529–544.
- Madeira, A.; Ohman, E.; Nilsson, A.; Sjögren, B.; Andrén, P.E.; Svenningsson, P. Coupling surface plasmon resonance to mass spectrometry to discover novel protein-protein interactions. Nat. Protoc. 2009, 4, 1023–1037.
- Boozer, C.; Kim, G.; Cong, S.; Guan, H.; Londergan, T. Looking towards label-free biomolecular interaction analysis in a high-throughput format: A review of new surface plasmon resonance technologies. Curr. Opin. Biotechnol. 2006, 17, 400–405.
- Thompson, P.M.; Beck, M.R.; Campbell, S.L. Protein-Protein Interactions, of the Series Methods in Molecular; Springer: New York, NY, USA, 2015; Volume 1278, pp. 267–279.
- Ranjbar, B.; Gill, P. Circular dichroism techniques: Biomolecular and nanostructural analyses—A review. Chem. Biol. Drug. Des. 2009, 74, 101–120.
- Some, D. Light-scattering-based analysis of biomolecular interactions. Biophys. Rev. 2013, 5, 147–158.
- Laue, T. Analytical ultracentrifugation: A powerful ‘new’ technology in drug discovery. Drug Discov. Today Technol. 2004, 1, 309–315.
- Velazquez-Campoy, A.; Leavitt, S.A.; Freire, E. Characterization of protein-protein interactions by isothermal titration calorimetry. Methods Mol. Biol. 2015, 1278, 183–204.
- Wienken, C.J.; Baaske, P.; Rothbauer, U.; Braun, D.; Duhr, S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 2010, 1, 100.
- Selvin, P.R. The renaissance of fluorescence resonance energy transfer. Nat. Struct. Biol. 2000, 7, 730–734.
- Pfleger, K.D.; Eidne, K.A. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods 2006, 3, 165–174.
- Yasgar, A.; Jadhav, A.; Simeonov, A.; Coussens, N.P. AlphaScreen-based assays: Ultra-high-throughput screening for small-molecule inhibitors of challenging enzymes and protein-protein interactions. Methods Mol. Biol. 2016, 1439, 77–98.
- Remy, I.; Campbell-Valois, F.X.; Michnick, S.W. Detection of protein-protein interactions using a simple survival protein-fragment complementation assay based on the enzyme dihydrofolate reductase. Nat. Protoc. 2007, 2, 2120–2125.
- Beeckmans, S. Chromatographic methods to study protein-protein interactions. Methods 1999, 19, 278–305.
- Muronetz, V.I.; Sholukh, M.; Korpela, T. Use of protein-protein interactions in affinity chromatography. J. Biochem. Biophys. Methods 2001, 49, 29–47.
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317.
- Pande, J.; Szewczyk, M.M.; Grover, A.K. Phage display: Concept, innovations, applications and future. Biotechnol. Adv. 2010, 28, 849–858.
- Fields, S.; Song, O. A novel genetic system to detect protein-protein interactions. Nature 1989, 340, 245–246.
- Hamdi, A.; Colas, P. Yeast two-hybrid methods and their applications in drug discovery. Trends Pharmacol. Sci. 2012, 33, 109–118.
- Hall, D.A.; Ptacek, J.; Snyder, M. Protein microarray technology. Mech. Ageing Dev. 2007, 128, 161–167.
- Zhu, H.; Bilgin, M.; Bangham, R.; Hall, D.; Casamayor, A.; Bertone, P.; Lan, N.; Jansen, R.; Bidlingmaier, S.; Houfek, T.; et al. Global analysis of protein activities using proteome chips. Science 2001, 293, 2101–2105.
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71.
- Karas, M.; Hillenkamp, F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 1988, 60, 2299–2301.
- Bich, C.; Zenobi, R. Mass spectrometry of large complexes. Curr. Opin. Struct. Biol. 2009, 19, 632–639.
- Tanaka, K.; Waki, H.; Ido, Y.; Akita, S.; Yoshida, Y.; Yoshida, T.; Matsuo, T. Protein and polymer analyses up to m/z 100,000 by laser ionization time of flight mass spectrometry. Rapid Commun. Mass Spectrom. 1988, 2, 151–153.
- Glish, G.L.; Vachet, R.W. The basics of mass spectrometry in the twenty-first century. Nat. Rev. Drug Discov. 2003, 2, 140–150.
- Kukacka, Z.; Rosulek, M.; Strohalm, M.; Kavan, D.; Novak, P. Mapping protein structural changes by quantitative cross-linking. Methods 2015, 89, 112–120.
- Fischer, L.; Chen, Z.A.; Rappsilber, J. Quantitative cross-linking/mass spectrometry using isotope-labelled cross-linkers. J. Proteom. 2013, 88, 120–128.
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386.
- Chavez, J.D.; Schweppe, D.K.; Eng, J.K.; Bruce, J.E. In Vivo Conformational Dynamics of Hsp90 and Its Interactors. Cell Chem. Biol. 2016, 23, 716–726.
- Pearson, K.M.; Pannell, L.K.; Fales, H.M. Intramolecular cross-linking experiments on cytochrome c and ribonuclease A using an isotope multiplet method. Rapid Commun. Mass Spectrom. 2002, 16, 149–159.
- Müller, F.; Graziadei, A.; Rappsilber, J. Quantitative Photo-crosslinking Mass Spectrometry Revealing Protein Structure Response to Environmental Changes. Anal. Chem. 2019, 91, 9041–9048.
- Chen, Z.A.; Rappsilber, J. Quantitative cross-linking/mass spectrometry to elucidate structural changes in proteins and their complexes. Nat. Protoc. 2019, 14, 171–201.
- Chen, Z.; Fischer, L.; Tahir, S.; Bukowski-Wills, J.C.; Barlow, P.; Rappsilber, J. Quantitative cross-linking/mass spectrometry reveals subtle protein conformational changes. Wellcome Open Res. 2016, 1, 5.
- Chen, Z.A.; Rappsilber, J. Protein Dynamics in Solution by Quantitative Crosslinking/Mass Spectrometry. Trends Biochem. Sci. 2018, 43, 908–920.
- Kleiner, R.E.; Hang, L.E.; Molloy, K.R.; Chait, B.T.; Kapoor, T.M. A Chemical Proteomics Approach to Reveal Direct Protein-Protein Interactions in Living Cells. Cell Chem. Biol. 2018, 25, 110–120.e3.
- Yu, C.; Huszagh, A.; Viner, R.; Novitsky, E.J.; Rychnovsky, S.D.; Huang, L. Developing a Multiplexed Quantitative Cross-Linking Mass Spectrometry Platform for Comparative Structural Analysis of Protein Complexes. Anal. Chem. 2016, 88, 10301–10308.
- Müller, F.; Rappsilber, J. A protocol for studying structural dynamics of proteins by quantitative crosslinking mass spectrometry and data-independent acquisition. J. Proteom. 2020, 218, 103721.
- Zhou, X.Z.; Lu, K.P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 2016, 16, 463–478.
- Shaw, P.E. Peptidyl-prolyl isomerases: A new twist to transcription. EMBO Rep. 2002, 3, 521–526.
- Lin, W.; Bonin, M.; Boden, A.; Wieduwild, R.; Murawala, P.; Wermke, M.; Andrade, H.; Bornhäuser, M.; Zhang, Y. Peptidyl prolyl cis/trans isomerase activity on the cell surface correlates with extracellular matrix development. Commun. Biol. 2019, 2, 58.
- Chavez, J.D.; Bruce, J.E. Chemical cross-linking with mass spectrometry: A tool for systems structural biology. Curr. Opin. Chem. Biol. 2019, 48, 8–18.
- Keller, A.; Chavez, J.D.; Tang, X.; Bruce, J.E. Leveraging the entirety of the protein data bank to enable improved structure prediction based on cross-link data. J. Proteome Res. 2021, 20, 1087–1095.
- Chavez, J.D.; Mohr, J.P.; Mathay, M.; Zhong, X.; Keller, A.; Bruce, J.E. Systems structural biology measurements by in vivo cross-linking with mass spectrometry. Nat. Protoc. 2019, 14, 2318–2343.
- Keller, A.; Chavez, J.D.; Felt, K.C.; Bruce, J.E. Prediction of an upper limit for the fraction of interprotein cross-links in large-scale in vivo cross-linking studies. J. Proteome Res. 2019, 18, 3077–3085.
- Liu, F.; Lössl, P.; Scheltema, R.; Viner, R.; Heck, A. Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification. Nat. Commun. 2017, 8, 15473.
- Klykov, O.; Steigenberger, B.; Pektaş, S.; Fasci, D.; Heck, A.; Scheltema, R.A. Efficient and robust proteome-wide approaches for cross-linking mass spectrometry. Nat. Protoc. 2018, 13, 2964–2990.
- Demartino, G.N. Reconstitution of PA700, the 19S regulatory particle, from purified precursor complexes. Methods Mol. Biol. 2012, 832, 443–452.
- Oeffinger, M. Two steps forward-one step back: Advances in affinity purification mass spectrometry of macromolecular complexes. Proteomics 2012, 12, 1591–1608.
- Mishra, P.K.; Yoo, C.M.; Hong, E.; Rhee, H.W. Photo-crosslinking: An emerging chemical tool for investigating molecular networks in live cells. ChemBioChem 2020, 21, 924–932.
- Tang, X.; Bruce, J.E. Mass Spectrometry of Proteins and Peptides, of the Series Methods in Molecular; Lipton, M.S., Paša-Tolic, L., Walker, J.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 492, pp. 283–293.
- Guerrero, C.; Tagwerker, C.; Kaiser, P.; Huang, L. An integrated mass spectrometry-based proteomic approach: Quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol. Cell Proteom. 2006, 5, 366–378.
- Vasilescu, J.; Guo, X.; Kast, J. Identification of protein-protein interactions using in vivo cross-linking and mass spectrometry. Proteomics 2004, 4, 3845–3854.
- Van den Heuvel, R.H.; Heck, A.J. Native protein mass spectrometry: From intact oligomers to functional machineries. Curr. Opin. Chem. Biol. 2004, 8, 519–526.
- Benesch, J.L.; Robinson, C.V. Mass spectrometry of macromolecular assemblies: Preservation and dissociation. Curr. Opin. Struct. Biol. 2006, 16, 245–251.
- Sharon, M.; Robinson, C.V. The role of mass spectrometry in structure elucidation of dynamic protein complexes. Annu. Rev. Biochem. 2007, 76, 167–193.
- Synowsky, S.A.; Van den Heuvel, R.H.; Mohammed, S.; Pijnappel, P.W.; Heck, A.J. Probing genuine strong interactions and post-translational modifications in the heterogeneous yeast exosome protein complex. Mol. Cell Proteom. 2006, 5, 1581–1592.
- Yu, C.; Huang, L. Cross-linking mass spectrometry: An emerging technology for interactomics and structural biology. Anal. Chem. 2018, 90, 144–165.

