 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Meinolf Suttorp | + 4139 word(s) | 4139 | 2021-03-11 04:49:14 | | | |
| 2 | Peter Tang | Meta information modification | 4139 | 2021-03-12 01:59:21 | | |
Video Upload Options
CML BCR-ABL1 positive is an acquired clonal myeloproliferative hematological malignancy derived from an abnormal pluripotent bone marrow stem cell. The leukemic cell clone consistently is characterized by a specific cytogenetic anomaly the so-called Philadelphia (Ph1) chromosome representing a reciprocal chromosomal translocation t(9;22)(q34.1;q11.2) which generates the BCR-ABL1 fusion gene. Cryptic translocations -being invisible on banding chromosome preparations- or variant translocations involving other chromosomes may represent an obstacle when establishing a diagnosis of CML. The presence of the Ph1 chromosome or BCR-ABL1 sharply separates CML from other myeloproliferative neoplasms (MPNs) like essential thrombocytosis (ET), polycythemia vera (PV) and idiopathic (osteo)myelofibrosis (OMF/IMF). Notably, the detection of the Ph1 chromosome is not sufficiently specific to diagnose CML, as it is also found in acute lymphoblastic leukemia (2–5% of pediatric cases of ALL). The BCR-ABL1 is present in the bone marrow in all myeloid lineages as well as in some lymphoid cells. Whether endothelial cells of the bone marrow niche are BCR-ABL1 positive is a matter of debate. Morphologically, CML is characterized by a hypercellular bone marrow, an unregulated growth of myeloid cells (neutrophils, eosinophils, basophils and megakaryocytes) resulting in abnormally high level of morphologically terminally differentiated granulocytes, as well as myeloid precursor cells in the blood and is associated with splenic enlargement in >60% of affected children.
1. Introduction
For a long time, the rarity of chronic myeloid leukemia (CML) in minors has hampered the accumulation of in-depth knowledge from cases presenting in the first two decades of life. Before the introduction of tyrosine kinase inhibitors (TKI) for the treatment of CML, stem cell transplantation was the recommended therapeutic approach for young patients and data on pediatric CML were deposited at the EBMT and IBMTR registries [1][2][3]. Given the enormous improvement of therapeutic success achieved by TKIs, the interest in the long-term outcome of this novel treatment especially in pediatric patients resulted in foundation of the International Registry on Pediatric CML (IR-PCML) at Poitiers/France in the year 2010 [4]. Since then, the number of collaboration centers and in parallel of registered patients continuously has increased. As of today, data on more than 660 patients diagnosed with CML at a median age of 12 years (range 0–17 years) have been collected. The information depicted from a registry on a rare disease like CML in minors offers the enormous benefit to enable treating physicians to apply a uniform approach to diagnose and follow-up this leukemia.
2. Related Terminology and ICD Codes
Synonyms of CML BCR-ABL1 positive are listed in Table 1. These terms are based on the laboratory method used to establish the diagnosis (based on chromosomal analysis or on molecular technique) and the terms to describe the myeloproliferative characteristics in histological findings. The term “myeloid” should be used in the English literature on pediatric CML for the sake of brevity and replace the terms “granulocytic” or “myelogenous”. The term “Juvenile CML” should not be used at all to avoid any possibility of confusion and mixing-up pediatric CML with the completely different entity of juvenile myelomonocytic leukemia (JMML) [5]. JMML is a unique pediatric disorder also different from chronic myelomonocytic leukemia (CMML) in adults [6][7][8].
Table 1. Synonyms of chronic myeloid leukemia (CML), BCR-ABL1 positive.
|
Synonym |
Abbreviation |
Comment |
|---|---|---|
|
CML, Philadelphia chromosome positive |
CML, Ph1 chromosome+ |
Nota bene: Ph1 stands for the word “Philadelphia” only * |
|
CML, t(9;22)(q34;q11) |
not applicable |
|
|
chronic granulocytic leukemia, BCR-ABL1 |
CML, BCR-ABL1 |
The abbreviation “CGL” should be avoided |
|
chronic granulocytic leukemia, Philadelphia chromosome positive |
CML, Ph1 chromosome+ |
The abbreviation “CGL” should be avoided |
|
chronic granulocytic leukemia, t(9;22)(q34;q11) |
CML, t(9;22)(q34;q11) |
The abbreviation “CGL” should be avoided |
|
chronic myelogenous leukemia, BCR-ABL1 positive |
CML, BCR-ABL1+ |
- |
|
chronic myelogenous leukemia, Philadelphia-chromosome positive |
CML, Ph1 chromosome+ |
- |
|
chronic myelogenous leukemia, t(9;22)(q34;q11) |
CML, t(9;22)(q34;q11) |
- |
* The abbreviations”, “Ph1”, “Ph-1”, “Ph 1” standing for “Philadelphia” can be found in the older literature and should not be used any longer.
The WHO’s system for International Classification of Diseases (ICD) in the present version of ICD11 recommends the codes as listed in Table 2 for categorization of CML. These codes do also apply in children with CML.
Table 2. Categorization of CML by the current codes of the International Classification of Diseases (ICD-11).
|
ICD-11 Code |
Category of CML |
Comment |
|---|---|---|
|
2B33.2 |
Chronic myeloid leukemia, not elsewhere classified |
only to be designated in cases with incomplete diagnostics |
|
2A20.0Y |
Other specified chronic myeloid leukemia, BCR-ABL1-positive |
e.g., CML, BCR-ABL1-positive, in complete remission |
|
2A20.0Z |
Chronic myeloid leukemia, BCR-ABL1-positive, unspecified |
e.g., no information on the phase of CML |
|
2A20.00 |
Chronic myeloid leukemia with blast crisis |
- |
|
2A20.01 |
Chronic myeloid leukemia, Philadelphia (Ph1) chromosome positive |
- |
|
2A20.02 |
Chronic myeloid leukemia, t(9:22)(q34;q11) |
- |
3. Staging and Classification of CML by Phases
Historically, based on the quantity of blasts, CML is categorized into three progressive phases driving the aggressiveness of the disease.
-
- Chronic phase (CML-CP) is the most common, indolent clinical stable phase of CML lasting for several years. The myeloid cells are differentiated, with less than 10% of blast cells present in the bone marrow. The response to therapy is excellent.
-
- If untreated, CML-CP usually progresses to the accelerated phase (CML-AP). The cells multiply aggressively and the blast cells increase to 10–19%. Additional chromosomal aberrations beside the Ph+ may be detectable [9]. The response to therapy becomes poorer.
-
- From CML-AP the leukemia progresses to a blastic phase (CML-BP) which is indistinguishable from acute leukemia exhibiting >20% (or ≥30%, see below) of bone marrow blasts of either myeloid or lymphoid immunophenotype. The response to therapy is very poor.
Logically the identification of the phase of CML forms the basis for treatment planning. However, the quantitative morphological criteria as established by the WHO [10][11] and the European LeukemiaNet (ELN) [12] as listed in Table 3 differ for CML-AP and CML-BP. For example, the WHO-recommended criteria for CML-BP are ≥20% of blast cells in blood or bone marrow, extramedullary blast proliferation, or large foci or clusters of blasts in the bone marrow biopsy while the ELN threshold is set to 30% of blasts. In adults, the borderline range has a clinical impact as in a comparative analysis, adult patients who had a blast percentage of 20–29% which is considered CML-BP according to the WHO classification, had a significantly better response rate (21% vs. 8%) and 3-year survival rate (42% vs. 10%) compared with patients who had blasts ≥ 30% [13].
Table 3. Comparison of the criteria established by the ELN and the WHO for definition of the phase of CML.
|
Definition as Used by |
||
|---|---|---|
|
Phase |
European LeukemiaNet (ELN) |
World Health Organization (WHO) |
|
CML CP |
|
|
|
CML-AP |
|
|
|
CML-BP |
|
|
In the TKI era, treatment response may also be used to classify CML-AP (provisional WHO definition) [10] such as:
-
hematologic resistance to the first TKI (or failure to achieve a complete hematologic response to the first TKI) or
-
any hematological, cytogenetic, or molecular indications of resistance to 2 sequential TKIs or
-
occurrence of 2 or more mutations in BCR-ABL1 during TKI therapy
The ELN criteria have been recommended previously [14] by the international Berlin-Frankfurt-Muenster (BFM) group also for pediatric CML as these criteria have been used in the majority of randomized clinical trials when treating CML in adults with TKI. Guidelines recently published by the Children’s Oncology Group from North America [15] are using the criteria of the National Comprehensive Cancer Network (NCCN) [16] which are derived from the WHO criteria.
In addition, it should be noticed that the introduction of new treatments could change the boundaries between CP, AP, and BC, and modify to some extent the classic subdivision of CML into three phases. Both classification systems agree on that independently from the proportion of blasts any extramedullary infiltration of organs beside liver and spleen in CML must be classified as blastic phase.
4. Subtypes of CML
One large breakpoint region (approx. 200 kb) is found in the ABL1 gene on chromosome 9q34 whereas three breakpoint regions are present in the so-called breakpoint cluster region (BCR) gene on chromosome 22q11 (Figure 1). Like in adults, in pediatric CML the vast majority of breakpoints cluster in a small region of the major (M-bcr) breakpoint while alternate, less common breakpoints cluster in the minor bcr (m-bcr) and very rarely further upstream in the BCR gene, in the so-called micro bcr (µ-bcr) [17][18][19]. M-bcr, m-bcr, and µ-bcr are associated with the p190, p210 and p230 BCR–ABL1 fusion proteins, respectively [20]. These three well-defined breakpoint regions in the BCR gene can produce at least eight different m-RNA fusion transcripts (M-bcr, p210: e14a2, e13a2, e14a3, e13a3; m-bcr, p190: e1a2, e1a3; µ-bcr, p230: e19a2, e19a3) because of alternative splicing in the ABL1 gene (splicing to exon 2 or exon 3) and because the M-bcr consists of two intronic regions (intron 13 and intron 14) [20][21].

Figure 1. Gene breakpoints and resulting transcript types. Intronic breakpoints (vertical black arrows) of the ABL1 gene and intronic breakpoints of the BCR gene (black horizontal arrows) and the corresponding fusion proteins (Length of exons and introns not according to scale). Pseudoexons 1a and 1b on the ABL1-gene as well as pseudoexons 1a and 2a on the BCR-gene are spliced out. In CML, the most frequently observed M-bcr breakpoint and the less frequently and only rarely observed breakpoints m-bcr and µ-bcr, respectively, can produce eight m-RNA fusion transcripts and translated proteins because of alternate splicing of the ABL1-gene exon 2 and because of two internal breakpoints in intron 13 and intron 14 of the BCR-gene. Additional breakpoints (indicated by a dotted horizontal arrow in this cartoon) have been described rarely or only as single case.
In CML-CP, the transcript types e13a2 and e14a2 are present with a frequency of 95%. In a single center study on pediatric CML (N = 146 patients), a proportion of 38% patients harbored transcript type e13a2 and 36% transcript e14a2 while the remaining 26% patients expressed both transcripts due to alternate splicing [17]. In a worldwide analysis on patients of all ages (N = 45,503 patients) transcript e13a2 is detected more frequently in males (39.2%) than in females (36.2%) and correlates with age, decreasing from 39.6% in children and adolescents down to 31.6% in patients ≥ 80 years old [22]. In addition, several very rare BCR–ABL1 variant fusion genes (resulting in the p195, p200 and p225 BCR–ABL1 fusion proteins; fusion transcripts e6a2, e8a2, and e18a2, respectively) have been detected -partly in single cases [21][22][23][24][25].
5. Differential Diagnosis
If CML is suspected but the Ph1 chromosome and BCR-ABL1 fusion transcript are absent, non-malignant disorders with a clinical and hematological picture mimicking CML need to be excluded first. Leukemoid reaction (LR) is the major differential diagnosis of CML in patients presenting with a leukocyte count in the range of 50,000 cells/µL and significant increase in mature neutrophils with a marked shift to the left [26]. Laboratory findings like toxic granulocytic vacuolation, Döhle’s bodies in the granulocytes, absence of basophilia, and a normal or increased leukocyte alkaline phosphatase (LAP) score separate the LR from CML [27]. Basophils are normal in LR and splenomegaly is an unusual finding. Taking carefully the clinical history and physical examination is suggestive of the origin of the LR, which is very heterogeneous comprising infections (especially S. aureus, S. pneumoniae), inflammatory syndromes (e.g., glomerulonephritis), malignancies, drugs (corticosteroids can cause a short-lasting extreme left-shifted neutrophilia), intoxications (liver failure), severe hemorrhage, and acute hemolysis [28].
Without karyotyping, CML may be more difficult to differentiate from the other classical myeloproliferative neoplasms (MPNs) occurring in adults which are comparatively uncommon in children. Polycythemia vera (PV) with associated iron deficiency (e.g., teenage girls with hypermenorrhagia), which causes normal hemoglobin and hematocrit values, can manifest with leukocytosis and thrombocytosis. Isolated megakaryocytic hyperplasia can be seen in Essential Thrombocythemia (ET) with marked thrombocytosis and splenomegaly. Such patients usually have a normal or increased LAP score, a WBC count less than 25,000/µL, and no Ph1 abnormality. However, concerning the typical mutations a lower incidence of mutation JAK2-V617F has been reported in childhood ET and PV, and fewer CALR mutations were found in children with ET [29]. Primary (osteo)myelofibrosis (PMF) also termed idiopathic myelofibrosis (IMF) is extremely rare in children [30], although sporadic childhood cases with PMF/IMF have been described [31]. Compared to adults, phenotypic differences appear to exist in children with PMF/IMF which are typically also found in pediatric CML, such as a frequent presence of marrow eosinophilia, only a low degree of marrow collagen fibrosis, the absence of significant osteosclerosis and megakaryocytic dysplasia with hypolobulated megakaryocytes with hyperchromatic nuclei and micromegakaryocytes. Notably, in none of 40 cases pediatric cases with IMF reported worldwide a previously described mutation, such as JAK2-V617F has been identified [32].
Virtually all cases of pediatric Myelodysplastic Syndrome (MDS) present with pancytopenia involving all three cell lineages, while single lineage cytopenia or macrocytosis may occasionally be the presenting findings [6]. Contrasting CML, leukocytosis is generally not a feature of MDS. Some patients may present with moderate hepatosplenomegaly but most have no organomegaly. Cytogenetic aberrations (monosomy 7, trisomy 8, 5q-, trisomy 21) are found in 55%–75% children with MDS. In childhood, the bone marrow may be hypo- or normocellular but rarely hypercellular. While small megakaryocytes are also found in CML, typical dysplastic features like macrocytic erythropoiesis, unusually large megakaryocytes, and dysgranulopoiesis point towards MDS but are not found in CML.
Juvenile Myelomonocytic Leukemia (JMML) typically manifests in infants and thereafter until the fifth year of life with declining incidence. Patients present with fever, infection, pallor, bleeding, hepato- or splenomegaly, lymphadenopathy, and skin rash [6]. The blood smear is characteristic showing uniformly elevated WBC count with absolute mo-nocytosis, anemia, and thrombocytopenia and is often more helpful in diagnosing than the BM morphology in which monocytosis is often only discrete. An additional finding not detected in CML is an increased HbF in patients with normal karyotype [5]. Mutations are found in the Ras signal transduction pathway downstream of the receptor in about 90% of patients; thus, JMML belongs to the group of diseases called RASopathies (Noonan syndrome, CBL-syndrome, NF1) [33].
Extremely rarely, pediatric patients may present with myeloid hyperplasia, which involves almost exclusively the neutrophil, eosinophil, or basophil cell lineage. These patients are described as having chronic neutrophilic, eosinophilic, or basophilic leukemia and do not have evidence of the Ph1 chromosome or the BCR-ABL1 gene. The World Health Organization defines MPD with eosinophilia and constitutively activated platelet-derived growth factor receptor-α, or -β, or fibroblast growth factor receptor 1 as a distinct category [34]. Occasionally, these myeloid neoplasms present in the first years of life with leukocytosis and organomegaly, and thus need to be differentiated from JMML or CML with increased eosinophils [5].
6. Epidemiology
CML usually presents at a median age of 60 years in Caucasians, but at younger age (35–45 years) in Asians [35][36][37]. It is rare in children contributing only 2–3% of all pediatric leukemia cases. The global incidence rate of CML is 15/1,000,000 per year with a male to female ratio of 1.34 while the age-adjusted incidence rate for the age group <18 years is 1.0 per 1,000,000 [38]. In the first three years of life pediatric CML is extremely rare [39]. The SEERS database showing pooled data on all myeloproliferative diseases at childhood age lists a continuously increasing incidence rate from 0.7 cases in the age group 1 to 4 years up to 4.3 cases per 1,000,000 at 15 to 19 years (Table 4). From the authors’ experience, myeloproliferative diseases besides CML like essential thrombocytosis, polycythemia vera and myelofibrosis are more than 10-fold less frequent than CML in the first two decades of life. Therefore, the SEERS data give a rather detailed impression on the continuous increase in the incidence of CML especially in the 2nd decade of life.
Table 4. Age adjusted and age specific incidence rates (per 1,000,000 children) of chronic myeloproliferatice diseases (all races, males and females) depicted from the SEERS database listing data from the USA in the years 2011–2015. [40].
|
Age (Years) |
0–14 |
0–19 |
<1 |
1–4 |
5–9 |
10–14 |
15–19 |
|---|---|---|---|---|---|---|---|
|
Chronic Myeloproliferative Diseases |
1.4 |
2.1 |
- |
0.7 |
1.0 |
2.1 |
4.3 |
7. Etiology
Predisposing factors to pediatric CML are not known. Ionizing radiation is considered a rare risk factor in adults. The maximum increase in CML incidence was observed in atomic bomb survivors in Hiroshima after a median time of 6 years, however, not after the Chernobyl nuclear power plant accident. Probably only exposure to higher radiation doses causes CML. Following irradiation and/or chemotherapy applied in the context of the treatment of a malignancy—mostly Hodgkin and Non-Hodgkin lymphomas—CML has been observed as secondary malignancy rarely in some adult and pediatric cases [41][42][43][44]. As the incidence of pediatric CML is not increased in healthy siblings, and especially not in twin pairs with one child affected from CML, genetic factors are of greater importance in the etiology of CML [45][46][47][48]. The role of mutated so-called myeloid “driver” genes is increasingly getting into the focus in pediatric CML [49][50].
8. Pathogenesis
The acquisition of BCR-ABL1 in a hematopoietic stem cell drives its transformation to become a leukemic stem cell (LSC). The fusion protein BCR-ABL1 represents a constitutive active tyrosine kinase considered to be the pathogenic driver capable of initiating and maintaining the disease. BCR-ABL1 activates a number of oncogenic signaling pathways, including PI3K/AKT/mTOR, RAS/RAF/MEK/ERK, and JAK/STAT [51]. However, numerous papers have described that the BCR-ABL1 oncogene does not operate alone when driving disease emergence, maintenance, and progression [52][53]. In children with CML, the myeloid driver mutation ASXL1 is found more frequently than in adult CML [49].
Additional biological differences in adult CML comprise a single breakpoint cluster within the first centromeric 1.5 kb of the BCR gene, whereas in pediatric CML there is a bimodal breakpoint distribution which is similar to adult Ph+ ALL harboring the less frequently observed M-BCR rearrangement [54]. In pediatric CML the bimodal breakpoint distribution in the BCR gene changes to the adult pattern at the age of 13 years, probably in association with the onset of puberty (Figure 2). For so far unknown reasons, the adult type breakpoint distribution pattern at prepubertal age is found more frequently in girls, but not in boys. We hypothesize that there are uncharacterized sex differences in the non-coding regions of the genome on which changes in the sex hormone blood levels starting at puberty exert an influence.

Figure 2. Illustration of the genomic DNA breakpoint distribution in the BCR gene and Kernel density analysis. For details see reference [54]. (A) When the pattern in 102 pediatric patients with CML is compared to 308 adult patients with CML as reported in the literature, pediatric CML shows a bimodal distribution which is also found in Ph1 positive acute lymphoblastic leukemia. (B) The bimodal “ALL-type” breakpoint can be found more frequently in prepubertal children and (C) in the prepubertal cohort is detectable more frequently in boys. In the Kernel plots the gray lines denote the 5% and the dotted line the 95% confidence intervals.
In pediatric acute leukemias the origin of the malignant cell clone has been traced back to its development in utero by using dried blood spots archived at birth on Guthrie cards [55][56]. The first case of pediatric CML in which BCR-ABL1, but no blood abnormalities were present at birth was identified recently from an archived cord blood specimen retrospectively when the infant presented with CML-CP at the age of 6 months [57]. A high-sensitive technique using nested genomic DNA-based PCR with subject-specific PCR primers after characterizing the genomic breakpoints [54][58][59] identified a clonal burden of less than 1 in 10,000 leukocytes at birth. In this case, a mutation associated with pre-disposition to several myeloid cancers was ruled out based on a defined myeloid panel of 11 driver genes (ACD, ANKRD26, CEBPA, DDX41, ETV6, GATA2, RUNX1, SRP72, TERC, TERT, and TP53). If all cases of CML –like acute leukemias– would also take their origin already in utero the observed sex difference at puberty concerning the BCR breakpoint could hardly be explained.
BCR-ABL1 can also be detected –rarely but with age-dependent increase from newborn to older age– in blood specimen collected from healthy individuals [60]. Thus, the dogma of BCR-ABL1 representing the sole event initiating CML is challenged [61]. In all patients receiving long-term TKI-treatment, CML-LSCs persist as they are resistant to the effects of TKIs. Bone marrow microenvironment-generated signals, cell autonomous BCR-ABL1 kinase-independent genetic changes, and epigenetic alterations all contribute to: (i) persistence of a quiescent LSC reservoir, (ii) innate or acquired resistance to TKIs, and (iii) progression into the fatal blast crisis stage [62][63].
9. Clinical Features and Hematological Findings
There are no organ alterations protruding as signs specific for CML. The bone marrow and blood are generally involved in pediatric CML. Besides hypersplenism, CML may cause fever, infection, easy bleeding, mild normocytic anemia, fatigue, bone pain, or other unspecific symptoms [39][64][65]. Compared to adults, pediatric CML-CP presents with more aggressive clinical and biological features such as a higher proportion of patients exhibiting splenomegaly, a larger spleen size, and higher leukocyte and platelets counts [64][66][67][68].
More than 90% of all pediatric patients with CML are diagnosed in CML-CP. The proportion of children in advanced phases (CML-AP and CML-BP) represents only 7.5% of all patients according to the International Registry for Childhood CML [4][69]. Because of leukemic infiltrates the spleen is enlarged in 70% to 80% and the liver in 50% to 60% of pediatric patients in CML-CP [64][65][70]. In CML-BP, however, any tissue (lymph nodes, skin, soft tissue, bones, and CNS) may be infiltrated by blasts [71]. Solid extramedullary manifestations of CML historically are termed chloroma because of the greenish color caused by the presence of myeloperoxidase [72]. Significantly, any extramedullary organ infiltration (except of spleen, liver, or retinal infiltration) results in upstaging a patient from CML-CP or CML-AP to CML-BP. CNS infiltration is not seen in CML-CP but the diagnosis of CML-BP requires i.th. prophylactic treatment [14][73].
CML is diagnosed in one third of pediatric patients incidentally when a blood count is performed to clarify other medical conditions [65][74]. Compared to adult CML, the mean leukocyte count in pediatric CML at diagnosis is more than four-fold higher (60 × 109 cells/L versus 240 × 109 cells/L) [65][66][74][75]. Therefore, once anticoagulated blood from pediatric patients with CML is allowed to separate into cellular elements and plasma, a broad “buffy coat” (“leukocrit”) overtopping the size of the hematocrit is usually visible. Mild normocytic, normochromic anemia (median Hb 10.4 g/dL) is present at diagnosis in 60% pediatric patients in CML-CP [64][65].
The WBC differential usually shows granulopoietic cells at all stages of maturation, from myeloblasts to mature, morphologically normal granulocytes. This “pathological shift to the left” is typical for CML and as outlined in Figure 3 easily allows the separation from other conditions like reactive shift to the left caused by infections or from AML with presence of blasts but missing more mature granulopoietic cells (hiatus leucemicus). Neutrophil function in CML is normal or only mildly impaired. These mature granulocytes have decreased apoptosis, resulting in accumulation of long-lived cells with low or absent enzymatic activity, such as alkaline phosphatase. Basophils are usually elevated in the range of 5% to 10% in the peripheral blood and eosinophils may be mildly increased as well.
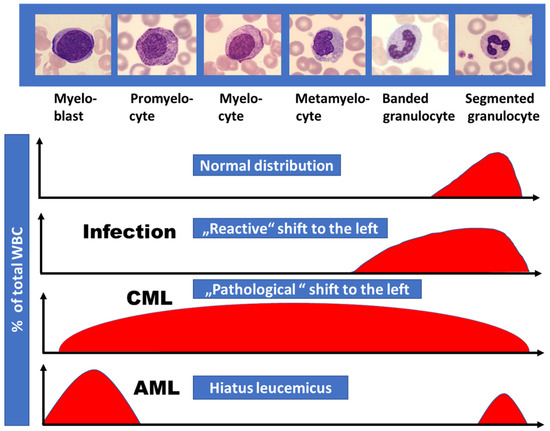
Figure 3. Proportion of immature granulopoietic cells (“shift to the left”) as observed in the differential white cell count in different diseases. Compared to the reactive shift to the left in bacterial infectious diseases, CML is characterized by the presence of more immature granulopoietic cells in addition including myeloblasts. In AML “hiatus leucemicus” (Latin, meaning “leukemic gap”) usually is observed exhibiting myeloblasts and mature granulocytes only.
Contrasting findings in acute leukemias, the platelet count is normal in CML-CP and even elevated above 500 × 109/L in half of the pediatric patients [76]. Thrombosis results extremely rarely from this alteration -instead mucocutaneous bleeding is observed in more than 10% children with elevated platelet counts. Bleeding is caused by a reduced plasma concentration of large Von Willebrand (VW) factor multimers, indicating a diagnosis of acquired VW-syndrome, which resolves after initiation of CML treatment. Platelet function abnormalities like Glanzmann thrombasthenia in CML at diagnosis have also been described in the literature [77].
During the accelerated phase of CML the proportion of immature cells increases and thrombocytopenia usually develops. Basophils may increase, and granulocyte maturation becomes defective.
References
- Champagne, M.A.; Capdeville, R.; Krailo, M.; Qu, W.; Peng, B.; Rosamilia, M.; Therrien, M.; Zoellner, U.; Blaney, S.M.; Bernstein, M.; et al. Imatinib mesylate (STI571) for treatment of children with Philadelphia chromosome-positive leukemia: Results from a Children’s Oncology Group phase 1 study. Blood 2004, 104, 2655–2660.
- Cwynarski, K.; Roberts, I.A.; Iacobelli, S.; van Biezen, A.; Brand, R.; Devergie, A.; Vossen, J.M.; Aljurf, M.; Arcese, W.; Locatelli, F.; et al. Stem cell transplantation for chronic myeloid leukemia in children. Blood 2003, 102, 1224–1231.
- Suttorp, M. Innovative approaches of targeted therapy for CML of childhood in combination with paediatric haematopoietic SCT. Bone Marrow Transplant. 2008, 42 (Suppl. S2), S40–S46.
- Suttorp, M.; Metzler, M.; Millot, F. Horn of plenty: Value of the international registry for pediatric chronic myeloid leukemia. World J. Clin Oncol. 2020, 11, 308–319.
- Niemeyer, C.M. JMML genomics and decisions. Hematol. Am. Soc. Hematol. Educ. Program. 2018, 2018, 307–312.
- Hasle, H. Myelodysplastic and myeloproliferative disorders of childhood. Hematol. Am. Soc. Hematol. Educ. Program. 2016, 2016, 598–604.
- Chang, T.Y.; Dvorak, C.C.; Loh, M.L. Bedside to bench in juvenile myelomonocytic leukemia: Insights into leukemogenesis from a rare pediatric leukemia. Blood 2014, 124, 2487–2497.
- Locatelli, F.; Niemeyer, C.M. How I treat juvenile myelomonocytic leukemia. Blood 2015, 125, 1083–1090.
- Millot, F.; Dupraz, C.; Guilhot, J.; Suttorp, M.; Brizard, F.; Leblanc, T.; Güneş, A.M.; Sedlacek, P.; De Bont, E.; Li, C.K.; et al. Additional cytogenetic abnormalities and variant t(9;22) at the diagnosis of childhood chronic myeloid leukemia: The experience of the International Registry for Chronic Myeloid Leukemia in Children and Adolescents. Cancer 2017, 123, 3609–3616.
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405.
- Vardiman, J.W.; Pierre, R.; Thiele, J.; Imbert, M.; Brunning, R.D.; Flandrin, G. Chronic myelogenous leukaemia. In World Health Organization Classification of Tumours: Pathology and Genetics—Tumours of Haematopoietic and Lymphoid Tissues; Jaffe, E.S., Harris, N.L., Stein, H., Vardiman, J.W., Eds.; IARC Press: Lyon, France, 2016; pp. 20–26.
- Baccarani, M.; Saglio, G.; Goldman, J.; Hochhaus, A.; Simonsson, B.; Appelbaum, F.; Apperley, J.; Cervantes, F.; Cortes, J.; Deininger, M.; et al. Evolving concepts in the management of chronic myeloid leukemia: Recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2006, 108, 1809–1820.
- Cortes, J.E.; Talpaz, M.; O’Brien, S.; Faderl, S.; Garcia-Manero, G.; Ferrajoli, A.; Verstovsek, S.; Rios, M.B.; Shan, J.; Kantarjian, H.M. Staging of chronic myeloid leukemia in the imatinib era: An evaluation of the World Health Organization proposal. Cancer 2006, 106, 1306–1315.
- De la Fuente, J.; Baruchel, A.; Biondi, A.; de Bont, E.; Dresse, M.F.; Suttorp, M.; Millot, F.; International BFM Group (iBFM) Study Group Chronic Myeloid Leukaemia Committee. Managing children with chronic myeloid leukaemia (CML): Recommendations for the management of CML in children and young people up to the age of 18 years. Br. J. Haematol. 2014, 167, 33–47.
- Athale, U.; Hijiya, N.; Patterson, B.C.; Bergsagel, J.; Andolina, J.R.; Bittencourt, H.; Schultz, K.R.; Burke, M.J.; Redell, M.S.; Kolb, E.A.; et al. Management of chronic myeloid leukemia in children and adolescents: Recommendations from the Children’s Oncology Group CML Working Group. Pediatr. Blood Cancer 2019, 66, e27827.
- NCCN Guidelines Version 2.Chronic Myeloid Leukemia. Available online: (accessed on 3 December 2020).
- Adler, R.; Viehmann, S.; Kuhlisch, E.; Martiniak, Y.; Röttgers, S.; Harbott, J.; Suttorp, M. Correlation of BCR/ABL transcript variants with patients’ characteristics in childhood chronic myeloid leukaemia. Eur. J. Haematol. 2009, 82, 112–118.
- Mughal, T.I.; Radich, J.P.; Deininger, M.W.; Apperley, J.F.; Hughes, T.P.; Harrison, C.J.; Gambacorti-Passerini, C.; Saglio, G.; Cortes, J.; Daley, G.Q. Chronic myeloid leukemia: Reminiscences and dreams. Haematologica 2016, 101, 541–558.
- Quintás-Cardama, A.; Cortes, J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630.
- Barnes, D.J.; Melo, J.V. Cytogenetic and molecular genetic aspects of chronic myeloid leukaemia. Acta Haematol. 2002, 108, 180–202.
- Melo, J.V. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood 1996, 88, 2375–2384.
- Baccarani, M.; Castagnetti, F.; Gugliotta, G.; Rosti, G.; Soverini, S.; Albeer, A.; Pfirrmann, M.; International BCR-ABL Study Group. The proportion of different BCR-ABL1 transcript types in chronic myeloid leukemia. An international overview. Leukemia 2019, 33, 1173–1183.
- Demehri, S.; Paschka, P.; Schultheis, B.; Lange, T.; Koizumi, T.; Sugimoto, T.; Branford, S.; Lim, L.C.; Kegel, T.; Martinelli, G.; et al. e8aBCR-ABL: More frequent than other atypical BCR-ABL variants? Leukemia 2005, 19, 681–684.
- Hochhaus, A.; Reiter, A.; Skladny, H.; Melo, J.V.; Sick, C.; Berger, U.; Guo, J.Q.; Arlinghaus, R.B.; Hehlmann, R.; Goldman, J.M.; et al. A novel BCR-ABL fusion gene (e6a2) in a patient with Philadelphia chromosome-negative chronic myelogenous leukemia. Blood 1996, 88, 2236–2240.
- Van der Velden, V.H.; Beverloo, H.B.; Hoogeveen, P.G.; Zwaan Ch, M. A novel BCR-ABL fusion transcript (e18a2) in a child with chronic myeloid leukemia. Leukemia 2007, 21, 833–835.
- Mandal, P.; Mukherjee, S.B. Leukemoid Reaction—A Tale of Years. Indian Pediatr. 2015, 52, 973–974.
- Sakka, V.; Tsiodras, S.; Giamarellos-Bourboulis, E.J.; Giamarellou, H. An update on the etiology and diagnostic evaluation of a leukemoid reaction. Eur. J. Intern. Med. 2006, 17, 394–398.
- Hoofien, A.; Yarden-Bilavski, H.; Ashkenazi, S.; Chodick, G.; Livni, G. Leukemoid reaction in the pediatric population: Etiologies, outcome, and implications. Eur. J. Pediatr. 2018, 177, 1029–1036.
- Karow, A.; Nienhold, R.; Lundberg, P.; Peroni, E.; Putti, M.C.; Randi, M.L.; Skoda, R.C. Mutational profile of childhood myeloproliferative neoplasms. Leukemia 2015, 29, 2407–2409.
- Sekhar, M.; Prentice, H.G.; Popat, U.; Anderson, D.; Janmohammed, R.; Roberts, I.; Britt, R.P. Idiopathic myelofibrosis in children. Br. J. Haematol. 1996, 93, 394–397.
- Ding, N.; Zhang, Z.; Yang, W.; Ren, L.; Zhang, Y.; Zhang, J.; Li, Z.; Zhang, P.; Zhu, X.; Chen, X.; et al. Transcriptome Analysis of Monozygotic Twin Brothers with Childhood Primary Myelofibrosis. Genom. Proteom. Bioinform. 2017, 15, 37–48.
- Mitton, B.; de Oliveira, S.; Pullarkat, S.T.; Moore, T.B. Stem cell transplantation in primary myelofibrosis of childhood. J. Pediatr. Hematol. Oncol. 2013, 35, e120–e122.
- Kratz, C.P.; Franke, L.; Peters, H.; Kohlschmidt, N.; Kazmierczak, B.; Finckh, U.; Bier, A.; Eichhorn, B.; Blank, C.; Kraus, C.; et al. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br. J. Cancer 2015, 112, 1392–1397.
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Arber, D.A.; Hasserjian, R.P.; Le Beau, M.M.; et al. (Eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC Press: Lyon, France, 2017.
- Malhotra, H.; Radich, J.; Garcia-Gonzalez, P. Meeting the Needs of CML Patients in Resource-Poor Countries. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 433–442.
- National Cancer Institute. Cancer Stat Facts: Leukemia—Chronic Myeloid Leukemia (CML). 2018. Available online: (accessed on 3 December 2020).
- Wang, A.; Wang, Y.; Yao, Y.; Xu, Z.; Zhou, L.; Wang, L.; Zhang, L.; Chen, Y.; Shen, Z.; Hu, J.; et al. Summary of 615 patients of chronic myeloid leukemia in Shanghai from 2001 to 2006. J. Exp. Clin. Cancer Res. 2010, 29, 20.
- Tanizawa, A. Optimal management for pediatric CML. Pediatrics Int. 2016, 58, 171–179.
- Meral Günes, A.; Millot, F.; Kalwak, K.; Lausen, B.; Sedlacek, P.; de Bruijn, C.A.M.; Dworzak, M.; de Moerloose, B.; Suttorp, M. Features and Outcome of Chronic Myeloid Leukemia at Very Young Age – Data from the International Pediatric CML Registry (I-CML-Ped Study). Ped Blood Cancer 2021, 68, e28706.
- Available online: (accessed on 1 August 2020).
- Alsop, S.; Sanger, W.G.; Elenitoba-Johnson, K.S.; Lim, M.S. Chronic myeloid leukemia as a secondary malignancy after ALK-positive anaplastic large cell lymphoma. Hum Pathol. 2007, 38, 1576–1580.
- Bauduer, F.; Ducout, L.; Dastugue, N.; Marolleau, J.P. Chronic myeloid leukemia as a secondary neoplasm after anti-cancer radiotherapy: A report of three cases and a brief review of the literature. Leuk Lymphoma 2002, 43, 1057–1060.
- Millett, R.; Aggarwal, A.; Tabbara, I.; Nassereddine, S. Chronic Myeloid Leukemia as Secondary Malignancy Following the Treatment of Hodgkin Lymphoma: A Case Series. Anticancer Res. 2019, 39, 4333–4335.
- Zahra, K.; Ben Fredj, W.; Ben Youssef, Y.; Zaghouani, H.; Chebchoub, I.; Zaier, M.; Badreddine, S.; Braham, N.; Sennana, H.; Khelif, A. Chronic myeloid leukemia as a secondary malignancy after lymphoma in a child. A case report and review of the literature. Onkologie 2012, 35, 690–693.
- Bizzozero, O.J., Jr.; Johnson, K.G.; Ciocco, A. Radiation-related leukemia in Hiroshima and Nagasaki, 1946-I. Distribution, incidence and appearance time. N. Engl. J. Med. 1966, 274, 1095–1101.
- Corso, A.; Lazzarino, M.; Morra, E.; Merante, S.; Astori, C.; Bernasconi, P.; Boni, M.; Bernasconi, C. Chronic myelogenous leukemia and exposure to ionizing radiation--a retrospective study of 443 patients. Ann. Hematol. 1995, 70, 79–82.
- Finch, S.C.; Linet, M.S. Chronic Leukaemias. Baillieres Clin Haematol. 1992, 5, 27–56.
- Radivoyevitch, T.; Jankovic, G.M.; Tiu, R.V.; Saunthararajah, Y.; Jackson, R.C.; Hlatky, L.R.; Gale, R.P.; Sachs, R.K. Sex differences in the incidence of chronic myeloid leukemia. Radiat. Environ. Biophys. 2014, 53, 55–63.
- Ernst, T.; Busch, M.; Rinke, J.; Ernst, J.; Haferlach, C.; Beck, J.F.; Hochhaus, A.; Gruhn, B. Frequent ASXL1 mutations in children and young adults with chronic myeloid leukemia. Leukemia 2018, 32, 2046–2049.
- Togasaki, E.; Takeda, J.; Yoshida, K.; Shiozawa, Y.; Takeuchi, M.; Oshima, M.; Saraya, A.; Iwama, A.; Yokote, K.; Sakaida, E.; et al. Frequent somatic mutations in epigenetic regulators in newly diagnosed chronic myeloid leukemia. Blood Cancer J. 2017, 7, e559.
- Deininger, M.W.; Goldman, J.M.; Melo, J.V. The molecular biology of chronic myeloid leukemia. Blood 2000, 96, 3343–3356.
- Branford, S.; Wang, P.; Yeung, D.T.; Thomson, D.; Purins, A.; Wadham, C.; Shahrin, N.H.; Marum, J.E.; Nataren, N.; Parker, W.T.; et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood 2018, 132, 948–961.
- Shanmuganathan, N.; Branford, S. The Hidden Pathogenesis of CML: Is BCR-ABL1 the First Event? Curr. Hematol. Malig. Rep. 2019, 14, 501–506.
- Krumbholz, M.; Karl, M.; Tauer, J.T.; Thiede, C.; Rascher, W.; Suttorp, M.; Metzler, M. Genomic BCR-ABL1 breakpoints in pediatric chronic myeloid leukemia. Genes Chromosomes Cancer 2012, 51, 1045–1053.
- Greaves, M.F.; Maia, A.T.; Wiemels, J.L.; Ford, A.M. Leukemia in twins: Lessons in natural history. Blood. 2003, 102, 2321–2333.
- Greaves, M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat. Rev. Cancer 2018, 18, 471–484.
- Abecasis, M.; Cross, N.C.P.; Brito, M.; Ferreira, I.; Sakamoto, K.M.; Hijiya, N.; Score, J.; Gale, R.P. Is cancer latency an outdated concept? Lessons from chronic myeloid leukemia. Leukemia 2020, 34, 2279–2284.
- Krumbholz, M.; Goerlitz, K.; Albert, C.; Lawlor, J.; Suttorp, M.; Metzler, M. Large amplicon droplet digital PCR for DNA-based monitoring of pediatric chronic myeloid leukaemia. J. Cell Mol. Med. 2019, 23, 4955–4961.
- Score, J.; Calasanz, M.J.; Ottman, O.; Pane, F.; Yeh, R.F.; Sobrinho-Simões, M.A.; Kreil, S.; Ward, D.; Hidalgo-Curtis, C.; Melo, J.V.; et al. Analysis of genomic breakpoints in p190 and pBCR-ABL indicate distinct mechanisms of formation. Leukemia 2010, 24, 1742–1750.
- Kuan, J.W.; Su, A.T.; Leong, C.F.; Osato, M.; Sashida, G. Systematic Review of Normal Subjects Harbouring BCR-ABLFusion Gene. Acta Haematol. 2020, 143, 96–111.
- Branford, S.; Kim, D.D.H.; Apperley, J.F.; Eide, C.A.; Mustjoki, S.; Ong, S.T.; Nteliopoulos, G.; Ernst, T.; Chuah, C.; Gambacorti-Passerini, C.; et al. Laying the foundation for genomically-based risk assessment in chronic myeloid leukemia. Leukemia 2019, 33, 1835–1850.
- Holyoake, T.L.; Vetrie, D. The chronic myeloid leukemia stem cell: Stemming the tide of persistence. Blood 2017, 129, 1160–1595.
- Perrotti, D.; Silvestri, G.; Stramucci, L.; Yu, J.; Trotta, R. Cellular and Molecular Networks in Chronic Myeloid Leukemia: The Leukemic Stem, Progenitor and Stromal Cell Interplay. Curr. Drug Targets 2017, 18, 377–388.
- Millot, F.; Traore, P.; Guilhot, J.; Nelken, B.; Leblanc, T.; Leverger, G.; Plantaz, D.; Bertrand, Y.; Bordigoni, P.; Guilhot, F. Clinical and biological features at diagnosis in 40 children with chronic myeloid leukemia. Pediatrics 2005, 116, 140–143.
- Suttorp, M.; Schulze, P.; Glauche, I.; Göhring, G.; von Neuhoff, N.; Metzler, M.; Sedlacek, P.; de Bont, E.S.J.M.; Balduzzi, A.; Lausen, B.; et al. Front-line imatinib treatment in children and adolescents with chronic myeloid leukemia: Results from a phase III trial. Leukemia 2018, 32, 1657–1669.
- Kalmanti, L.; Saussele, S.; Lauseker, M.; Proetel, U.; Müller, M.C.; Hanfstein, B.; Schreiber, A.; Fabarius, A.; Pfirrmann, M.; Schnittger, S.; et al. Younger patients with chronic myeloid leukemia do well in spite of poor prognostic indicators: Results from the randomized CML study IV. Ann. Hematol. 2014, 93, 71–80.
- Millot, F.; Guilhot, J.; Suttorp, M.; Güneş, A.M.; Sedlacek, P.; De Bont, E.; Li, C.K.; Kalwak, K.; Lausen, B.; Culic, S.; et al. Prognostic discrimination based on the EUTOS long-term survival score within the International Registry for Chronic Myeloid Leukemia in children and adolescents. Haematologica 2017, 102, 1704–1708.
- Suttorp, M.; Knöfler, R.; Deutsch, H.; Paul, F.; Tiebel, O.; Metzler, M.; Millot, F. High Platelet Counts, Thrombosis, Bleeding Signs, and Acquired Von Willebrand Syndrome at Diagnosis of Pediatric Chronic Myeloid Leukemia (Abstract). Blood 2019, 134 (Suppl. S1), 4152.
- Millot, F.; Maledon, N.; Guilhot, J.; Güneş, A.M.; Kalwak, K.; Suttorp, M. Favourable outcome of de novo advanced phases of childhood chronic myeloid leukaemia. Eur. J. Cancer. 2019, 115, 17–23.
- Hijiya, N.; Millot, F.; Suttorp, M. Chronic myeloid leukemia in children: Clinical findings, management, and unanswered questions. Pediatr. Clin North Am. 2015, 62, 107–119.
- Millot, F.; Facon, T.; Kerckaert, J.P.; Fenaux, P.; Lai, J.L.; Parent, M.; Bauters, F.; Jouet, J.P. Unusual recurrence of chronic myelogenous leukemia following bone marrow transplantation. Bone Marrow Transpl. 1991, 7, 393–395.
- Magdy, M.; Abdel Karim, N.; Eldessouki, I.; Gaber, O.; Rahouma, M.; Ghareeb, M. Myeloid Sarcoma. Oncol. Res. Treat. 2019, 42, 224–229.
- Meyran, D.; Petit, A.; Guilhot, J.; Suttorp, M.; Sedlacek, P.; De Bont, E.; Li, C.K.; Kalwak, K.; Lausen, B.; Culic, S.; et al. Lymphoblastic predominance of blastic phase in children with chronic myeloid leukemia (CML) treated with imatinib: A report from the I-CML-Ped Study. Eur. J. Cancer 2020, 137, 224–234.
- Millot, F.; Baruchel, A.; Guilhot, J.; Petit, A.; Leblanc, T.; Bertrand, Y.; Mazingue, F.; Lutz, P.; Vérité, C.; Berthou, C.; et al. Imatinib is effective in children with previously untreated chronic myelogenous leukemia in early chronic phase: Results of the French national phase IV trial. J. Clin. Oncol. 2011, 29, 2827–2832.
- Castagnetti, F.; Gugliotta, G.; Baccarani, M.; Breccia, M.; Specchia, G.; Levato, L.; Abruzzese, E.; Rossi, G.; Iurlo, A.; Martino, B.; et al. Differences among young adults, adults and elderly chronic myeloid leukemia patients. Ann. Oncol. 2015, 26, 185–192.
- Knöfler, R.; Lange, B.S.; Paul, F.; Tiebel, O.; Suttorp, M. Bleeding signs due to acquired von Willebrand syndrome at diagnosis of chronic myeloid leukaemia in children. Br. J. Haematol. 2020, 188, 701–706.
- Chauhan, R.; Sazawal, S.; Singh, K.; Ragesh RNair, R.; Chhikara, S.; Deka, R.; Chaubey, R.; Veetil, K.K.; Dange, P.; Mahapatra, M.; et al. Reversal of Glanzmann thrombasthenia platelet phenotype after imatinib treatment in a pediatric chronic myeloid leukemia patient. Platelets 2018, 29, 203–206.

