 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Annelies De Maré | + 1342 word(s) | 1342 | 2020-05-08 05:59:14 | | | |
| 2 | Nicole Yin | -15 word(s) | 1327 | 2020-10-29 08:44:34 | | |
Video Upload Options
Sclerostin, a 22-kDa glycoprotein that is mainly secreted by the osteocytes, is a soluble inhibitor of canonical Wnt signaling. This canonical Wnt signaling pathway, in addition to its function during embryogenesis, also plays a crucial role in adult tissue homeostasis by regulating the maintenance and differentiation of stem cells. In particular, this signaling cascade also exerts an important regulatory pathway in the differentiation of mesenchymal stem cells towards the osteoblast-lineage. Since sclerostin is an inhibitor of this signaling pathway, increased sclerostin concentrations will lead to an increased bone resorption and decreased bone formation. The cellular and molecular actions that are involved in this process, will be discussed in this review.
1. Introduction
Beta-catenin is the central regulatory player in the canonical Wnt signaling. Activation of this signaling cascade, by binding of the Wnt ligands to the Frizzled (Fz) receptor and Low-density Lipoprotein Receptor-related Protein 5/6 (LRP5/6) co-receptors, leads to inhibition of the β-catenin degradation complex. In this way, β-catenin can accumulate in the cytoplasm, and subsequently be translocated into the nucleus. In the nucleus, β-catenin functions as a coactivator of the transcription factors T-cell factor (TCF) and Lymphoid Enhancer-binding factor (LEF), thereby modifying gene transcription. It has been shown that the Wnt/β-catenin signaling cascade downregulates adipogenic differentiation by inhibiting the expression of Peroxisome Proliferator-Activated Receptor gamma (PPARγ) and CCAAT/Enhancer Binding Protein alpha (C/EBPα), both important adipogenic regulators, while stimulating Runt-related transcription factor 2 (Runx2) and Osterix, well-known inducers of osteogenesis [1][2]. The canonical Wnt signaling also stimulates osteoblast maturation and viability of osteoblasts and osteocytes. These cells then increase their production of osteoprotegerin (OPG) (a decoy receptor of Receptor Activator of Nuclear Factor Kappa-Β Ligand (RANKL)), by which osteoclast formation is inhibited.
To prevent excessive bone formation, several antagonists are produced amongst which is sclerostin. Mechanical unloading [3], low levels of serum parathyroid hormone (PTH) [4][5] and estrogen deficiency [6] trigger sclerostin production. As already mentioned above, in the bone, sclerostin is mainly produced by the osteocytes, the cells that reside within the bone matrix and comprise between 90%–95% of all bone cells. After its secretion, sclerostin will be anchored to the LRP4 receptor on the osteoblast membrane, by which sclerostin is retained in the bone compartment [7]. Sclerostin can also bind to LRP5/6, leading to receptor internalization and/or reduced availability of these co-receptors to Wnt ligands, which results in inhibition of the canonical Wnt signaling. This leads to (Figure 1):
2.Inhibition of Proliferation and Differentiation of Osteoprogenitor/Pre-Osteoblastic Cells, as Well as Decreased Activation of Mature Osteoblasts
Osteoblasts are derived from mesenchymal stems cells, which are multipotent progenitor cells that are able to differentiate into a variety of cell types (including osteoblasts, chondrocytes, adipocytes, smooth muscle cells [8] and endothelial cells [9]). Depending on the specific activation of signaling pathways (such as Wnt/β-catenin signaling) and transcription factors (such as Runx2 and osterix), mesenchymal cells will commit to the osteoblastic lineage. Inhibition of the canonical Wnt signaling by sclerostin therefore directly prevents the development of new osteoblasts. However, Thouverey and Caverzasio found that sclerostin not only functions by inhibiting canonical Wnt signaling, but also activates platelet-derived growth factor receptor signaling to inhibit osteoblast differentiation [10]. Sclerostin also inhibits the activity of mature osteoblasts, since osteocalcin, procollagen type 1 N-terminal Propeptide (P1NP) and bone-specific alkaline phosphatase (BsAP), all produced by the osteoblast and therefore considered indicators of osteoblastic activity, were increased after administration of romosozumab, an antibody directed against sclerostin [11].
3.Decreased Mineralization
Mineralization of newly formed bone is a dynamic process in which Small Integrin-Binding Ligand N-linked Glycoproteins (SIBLINGS), such as Matrix Extracellular Phosphoglycoprotein (MEPE), are involved [12]. A key characteristic of MEPE, and several other SIBLING proteins, is the presence of an Acidic Serine Aspartate-Rich MEPE-associated (ASARM) motif [13]. When cleaved by cathepsin B, the ASARM motif inhibits mineralization and phosphate uptake [14]. Cleavage of this ASARM motif can be prevented by Phosphate-regulating neutral Endopeptidase (PHEX), which binds to full-length MEPE [15][16]. Regulation of mineralization is therefore determined by the PHEX/MEPE ratio. Sclerostin is involved in the regulation of this PHEX/MEPE axis, by inducing the expression of ASARM-peptides (anti-mineralization) and downregulating PHEX (pro-mineralization) [17][18].
4. Increased Apoptosis of the Osteogenic Cells
Experiments in Sost knockout (KO) mice demonstrated decreased apoptosis of osteoblasts and osteocytes, while osteoblast activity was increased in these animals compared to wild type (WT) mice [19]. These results were confirmed by Chandra et al., showing that in a mouse model of radiation damage, the inhibition of sclerostin protected against apoptosis of osteoblasts [20]. Additionally, in human osteoblastic cells, sclerostin induced apoptosis by activating caspases 1, 3, 4 and 7, as well increasing the expression of the pro-apoptotic factor Bax [21].
5. Maintenance of Bone Lining Cells in the Quiescent State
Bone lining cells are found covering the bone surface. They are considered to be derived from previously active osteoblasts that did not undergo apoptosis or differentiated into osteocytes [22]. During normal physiology, active bone lining cells play a crucial role as coordinators of bone formation and resorption [23][24]. Regulation of the activity state of the bone lining cells—either remaining in the quiescent state or being reactivated to osteoblasts—at least in part, seems to be controlled by sclerostin. One study, performed in ovariectomized rats and cynomolgus monkeys, demonstrated that anti-sclerostin antibody treatment strongly reduced quiescent bone surfaces, whilst increasing bone surfaces actively involved in mineralization [25]. Another study in which mice were administered with an anti-sclerostin antibody, showed reactivation of the bone lining cells, as indicated by the increase in size and expression of osteocalcin, a marker of osteoblastic bone formation [26]. These results demonstrate that reactivation of bone lining cells could explain the rapid increase in osteoblast numbers on previously quiescent bone surfaces [26].
6. Regulation of Osteocyte Maturation and Osteocytic Osteolysis
Immature osteocytes, which are located in the osteoid or in close proximity of the bone surface, still share many morphological features with the osteoblast [27]. However, when the osteocytes becomes deeper embedded in the mineralized bone matrix, they change their morphology to become mature osteocytes [27]. This process of osteocyte maturation is triggered by matrix mineralization [27].
As discussed above, sclerostin is an inhibitor of matrix mineralization and therefore, could also be involved in the regulation of osteocyte maturation. This was demonstrated by Atkins et al., who found that, after treating human primary osteoblast cultures with sclerostin, cells increased their E11 expression (pre-osteocyte marker), while decreasing dentin matrix acidic phosphoprotein 1 (DMP1) and SOST expression (mature osteocyte markers) [17].
In the bone, sclerostin is produced by the mature osteocytes, where its expression is regulated by a wide variety of factors, including local cytokines [28], hormones such as PTH [5] and estrogen [29], and mechanical loading [30]. Under unloading conditions, sclerostin is produced by the osteocytes in order to regulate re-shaping of the osteocytic lacunae by ‘osteocytic osteolysis’ [31]. The proteins that are upregulated by sclerostin correspond to those produced by the osteoclasts, including carbonic anhydrase 2 [31], cathepsin K [32][31], tartrate-resistant acid phosphatase [32][31], and C-terminal collagen telopeptide [18]. This process of osteocytic osteolysis has important effects on bone physiology, not only by releasing calcium from the bone matrix, which is shown to be crucial during lactation [32], but also by affecting mechano-sensation and bone turnover (reviewed by Tsourdi et al. [33]).
7. Stimulation of Bone Resorption
Research has shown that the canonical Wnt/β-catenin signaling is a critical regulator of osteoclastogenesis [34][35][36]. During the inhibition of canonical Wnt signaling, the expression of OPG is decreased, thereby increasing the RANKL/OPG ratio, and, thus, bone resorption [37]. Wei et al. showed that, in response to RANKL, β-catenin in osteoclast precursors is downregulated, which is needed to allow the differentiation of osteoclast precursors towards mature osteoclasts [34]. Furthermore, Weivoda et al. demonstrated that osteoclast lineage cells express canonical Wnt receptors [36], indicating that sclerostin might have direct effects on osteoclast formation and maturation.
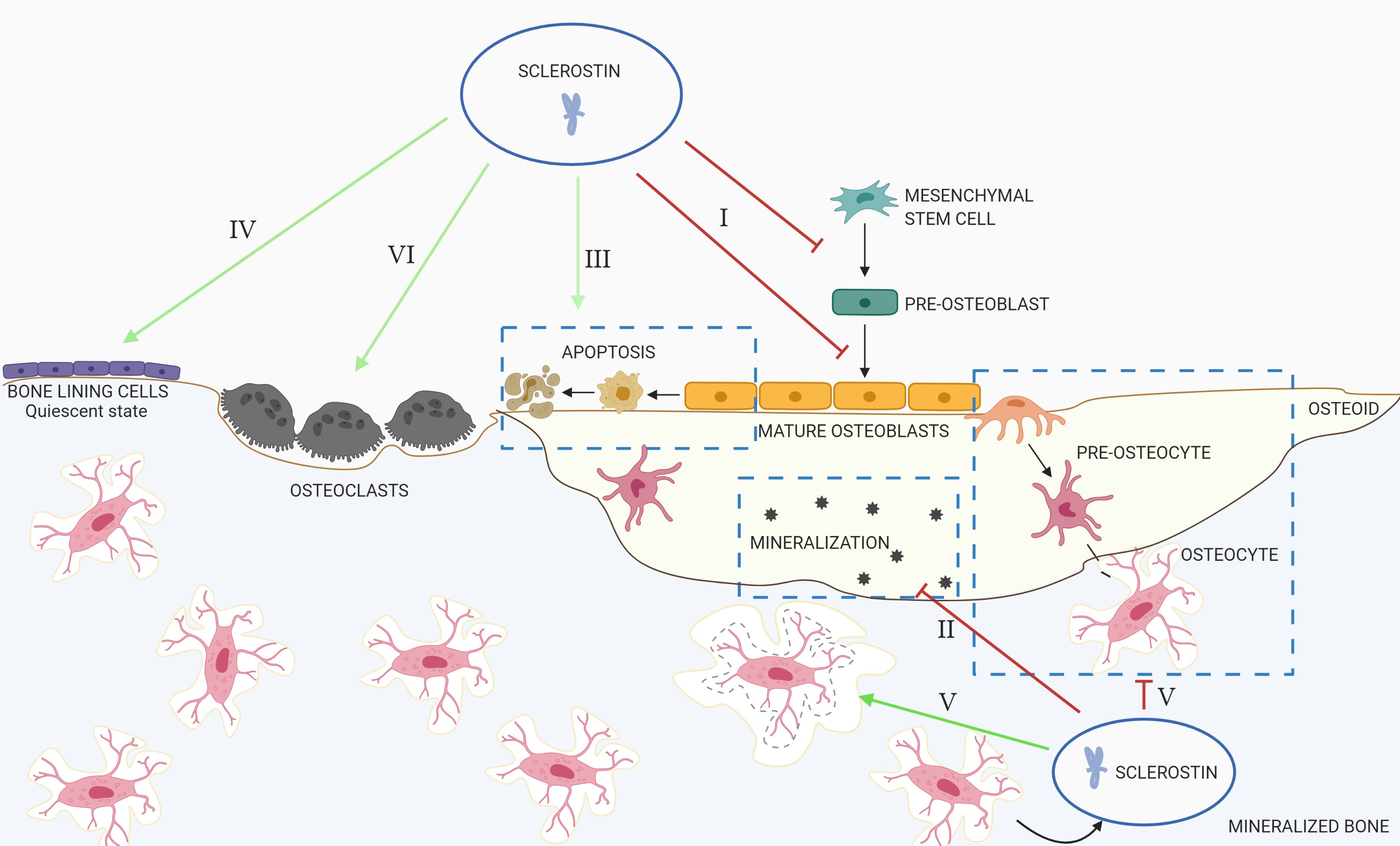
Figure 1. Overview of the actions of sclerostin in the bone. I: Inhibition of proliferation and differentiation of osteoprogenitor/pre-osteoblastic cells, as well as decreased activation of mature osteoblasts; II: decreased mineralization; III: increased apoptosis of the osteogenic cells; IV: maintenance of bone lining cells in their quiescent state; V: regulation of osteocyte maturation and osteocytic osteolysis; VI: stimulation of bone resorption.
References
- S. Kang; C. N. Bennett; I. Gerin; L. A. Rapp; Kurt D. Hankenson; Ormond MacDougald; Wnt Signaling Stimulates Osteoblastogenesis of Mesenchymal Precursors by Suppressing CCAAT/Enhancer-binding Protein and Peroxisome Proliferator-activated Receptor. Journal of Biological Chemistry 2007, 282, 14515-14524, 10.1074/jbc.m700030200.
- Christina N. Bennett; Kenneth A. Longo; Wendy S. Wright; Larry J. Suva; Timothy F. Lane; Kurt D. Hankenson; Ormond MacDougald; Regulation of osteoblastogenesis and bone mass by Wnt10b. Proceedings of the National Academy of Sciences 2005, 102, 3324-3329, 10.1073/pnas.0408742102.
- Alexander G. Robling; P. J. Niziolek; L. A. Baldridge; K. W. Condon; Matthew R. Allen; I. Alam; S. M. Mantila; J. Gluhak-Heinrich; Teresita Bellido; S. E. Harris; et al.C. H. Turner Mechanical Stimulation of Bone in Vivo Reduces Osteocyte Expression of Sost/Sclerostin. Journal of Biological Chemistry 2007, 283, 5866-5875, 10.1074/jbc.m705092200.
- Teresita Bellido; A. A. Ali; I. Gubrij; L. I. Plotkin; Q. Fu; C A O'brien; S. C. Manolagas; Robert L. Jilka; Chronic Elevation of Parathyroid Hormone in Mice Reduces Expression of Sclerostin by Osteocytes: A Novel Mechanism for Hormonal Control of Osteoblastogenesis. Endocrinology 2005, 146, 4577-4583, 10.1210/en.2005-0239.
- Hansjoerg Keller; Michaela Kneissel; SOST is a target gene for PTH in bone. Bone 2005, 37, 148-158, 10.1016/j.bone.2005.03.018.
- Koji Fujita; Matthew M. Roforth; Susan Demaray; Ulrike McGregor; Salman Kirmani; Louise K. McCready; James M. Peterson; Matthew T. Drake; David G. Monroe; Sundeep Khosla; et al. Effects of estrogen on bone mRNA levels of sclerostin and other genes relevant to bone metabolism in postmenopausal women.. The Journal of Clinical Endocrinology & Metabolism 2013, 99, E81-8, 10.1210/jc.2013-3249.
- Eveline Boudin; Timur Yorgan; Igor Fijałkowski; Stephan Sonntag; Ellen Steenackers; Gretl Hendrickx; Silke Peeters; Annelies De Maré; Benjamin A. Vervaet; Anja Verhulst; et al.Geert MortierPatrick D'haeseThorsten SchinkeWim Van Hul The Lrp4 R1170Q Homozygous Knock-In Mouse Recapitulates the Bone Phenotype of Sclerosteosis in Humans. Journal of Bone and Mineral Research 2017, 32, 1739-1749, 10.1002/jbmr.3160.
- Zhaodi Gong; Laura E. Niklason; Small‐diameter human vessel wall engineered from bone marrow‐derived mesenchymal stem cells (hMSCs). The FASEB Journal 2008, 22, 1635-1648, 10.1096/fj.07-087924.
- Joachim Oswald; Sabine Boxberger; Birgitte Jørgensen; Silvia Feldmann; Gerhard Ehninger; Martin Bornhäuser; Carsten Werner; Mesenchymal Stem Cells Can Be Differentiated Into Endothelial Cells In Vitro. STEM CELLS 2004, 22, 377-384, 10.1634/stemcells.22-3-377.
- Cyril Thouverey; Joseph Caverzasio; Sclerostin inhibits osteoblast differentiation without affecting BMP2/SMAD1/5 or Wnt3a/β-catenin signaling but through activation of platelet-derived growth factor receptor signaling in vitro. BoneKEy Reports 2015, 4, 757, 10.1038/bonekey.2015.126.
- Desmond Padhi; Graham Jang; Brian Stouch; Liang Fang; Edward Posvar; Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. Journal of Bone and Mineral Research 2010, 26, 19-26, 10.1002/jbmr.173.
- Aline Martin; Valentin David; Jennifer S. Laurence; Patricia M. Schwarz; Eileen Lafer; Anne-Marie Hedge; Peter S.N. Rowe; Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-peptide(s) are directly responsible for defective mineralization in HYP.. Endocrinology 2007, 149, 1757-72, 10.1210/en.2007-1205.
- P S N Rowe; Priyal A De Zoysa; Rong Dong; Huei Rong Wang; Kenneth E White; Michael Econs; Claudine L Oudet; MEPE, a New Gene Expressed in Bone Marrow and Tumors Causing Osteomalacia. Genomics 2000, 67, 54-68, 10.1006/geno.2000.6235.
- Peter S.N. Rowe; The wrickkened pathways of FGF23, MEPE and PHEX.. Critical Reviews in Oral Biology & Medicine 2004, 15, 264-81.
- Rong Guo; Peter S N Rowe; Shiguang Liu; Leigh G Simpson; Zhousheng Xiao; L. Darryl Quarles; Inhibition of MEPE cleavage by Phex.. Biochemical and Biophysical Research Communications 2002, 297, 38-45, 10.1016/s0006-291x(02)02125-3.
- Peter S.N. Rowe; Y. Kumagai; G. Gutierrez; I.R. Garrett; R. Blacher; D. Rosen; J. Cundy; S. Navvab; D. Chen; M.K. Drezner; et al.L. Darryl QuarlesG.R. Mundy MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone 2004, 34, 303-319, 10.1016/j.bone.2003.10.005.
- Gerald J. Atkins; Peter S Rowe; Hui P Lim; Katie J Welldon; Renee Ormsby; Asiri R Wijenayaka; Lesya Zelenchuk; Andreas Evdokiou; David M Findlay; Sclerostin is a locally acting regulator of late-osteoblast/preosteocyte differentiation and regulates mineralization through a MEPE-ASARM-dependent mechanism. Journal of Bone and Mineral Research 2011, 26, 1425-1436, 10.1002/jbmr.345.
- Masakazu Kogawa; Kamarul A Khalid; Asiri R. Wijenayaka; Renee T. Ormsby; Andreas Evdokiou; Paul H Anderson; David M Findlay; Gerald J. Atkins; Recombinant sclerostin antagonizes effects of ex vivo mechanical loading in trabecular bone and increases osteocyte lacunar size.. American Journal of Physiology-Cell Physiology 2017, 314, C53-C61, 10.1152/ajpcell.00175.2017.
- Carola Krause; Olexandr Korchynskyi; Karien De Rooij; Stella E. Weidauer; David J. J. De Gorter; Rutger L. Van Bezooijen; Sarah Hatsell; Aris N. Economides; Thomas D. Mueller; Clemens W. G. M. Löwik; et al.Peter Ten Dijke Distinct Modes of Inhibition by Sclerostin on Bone Morphogenetic Protein and Wnt Signaling Pathways*. Journal of Biological Chemistry 2010, 285, 41614-41626, 10.1074/jbc.M110.153890.
- Abhishek Chandra; Tiao Lin; Tiffany Young; Wei Tong; Xiaoyuan Ma; Wei-Ju Tseng; Ina Kramer; Michaela Kneissel; Michael A Levine; Yejia Zhang; et al.Keith CengelX. Sherry LiuLing Qin Suppression of Sclerostin Alleviates Radiation-Induced Bone Loss by Protecting Bone-Forming Cells and Their Progenitors Through Distinct Mechanisms.. Journal of Bone and Mineral Research 2016, 32, 360-372, 10.1002/jbmr.2996.
- May Kung Sutherland; James C. Geoghegan; Changpu Yu; Eileen Turcott; John E. Skonier; David G. Winkler; John A. Latham; Sclerostin promotes the apoptosis of human osteoblastic cells: a novel regulation of bone formation. Bone 2004, 35, 828-835, 10.1016/j.bone.2004.05.023.
- Morten A. Karsdal; Lykke Larsen; Michael T. Engsig; Henriette Lou; Mercedes Ferreras; André Lochter; Jean-Marie Delaisse; Niels T. Foged; Matrix Metalloproteinase-dependent Activation of Latent Transforming Growth Factor-β Controls the Conversion of Osteoblasts into Osteocytes by Blocking Osteoblast Apoptosis. Journal of Biological Chemistry 2002, 277, 44061-44067, 10.1074/jbc.m207205200.
- Orit Kollet; Ayelet Dar; Shoham Shivtiel; Alexander Kalinkovich; Tsvee Lapidot; Yejezkel Sztainberg; Melania Tesio; Robert M. Samstein; Polina Goichberg; Asaf Spiegel; et al.Ari ElsonTsvee Lapidot Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nature Medicine 2006, 12, 657-664, 10.1038/nm1417.
- V. Everts; Jean-Marie Delaisse; W. Korper; D. C. Jansen; W. Tigchelaar-Gutter; P. Saftig; Wouter Beertsen; The Bone Lining Cell: Its Role in Cleaning Howship's Lacunae and Initiating Bone Formation. Journal of Bone and Mineral Research 2002, 17, 77-90, 10.1359/jbmr.2002.17.1.77.
- Michael S Ominsky; Qing-Tian Niu; Chaoyang Li; Xiaodong Li; Hua Zhu Ke; Tissue-Level Mechanisms Responsible for the Increase in Bone Formation and Bone Volume by Sclerostin Antibody. Journal of Bone and Mineral Research 2014, 29, 1424-1430, 10.1002/jbmr.2152.
- Sang Wan Kim; Yanhui Lu; Elizabeth A. Williams; Forest Lai; Ji Yeon Lee; Tetsuya Enishi; Deepak H. Balani; Michael S. Ominsky; Hua Zhu Ke; Henry M. Kronenberg; et al.Marc N. Wein Sclerostin Antibody Administration Converts Bone Lining Cells Into Active Osteoblasts.. Journal of Bone and Mineral Research 2017, 32, 892-901, 10.1002/jbmr.3038.
- Kazuharu Irie; Sadakazu Ejiri; Yasunori Sakakura; Toru Shibui; Toshihiko Yajima; Matrix Mineralization as a Trigger for Osteocyte Maturation. Journal of Histochemistry & Cytochemistry 2008, 56, 561-567, 10.1369/jhc.2008.950527.
- D. C. Genetos; Clare E. Yellowley; Gabriela G. Loots; Prostaglandin E2 Signals Through PTGER2 to Regulate Sclerostin Expression. PLOS ONE 2011, 6, e17772, 10.1371/journal.pone.0017772.
- David G. Winkler; May Kung Sutherland; James C. Geoghegan; Changpu Yu; Trenton Hayes; John E. Skonier; Diana Shpektor; Mechtild Jonas; Brian R. Kovacevich; Karen Staehling‐Hampton; et al.Mark ApplebyMary E. BrunkowJohn A. Latham Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. The EMBO Journal 2003, 22, 6267-6276, 10.1093/emboj/cdg599.
- Masakazu Kogawa; Asiri R Wijenayaka; Renee T Ormsby; Gethin Thomas; Paul Anderson; Lynda F Bonewald; David M Findlay; Gerald J. Atkins; Sclerostin Regulates Release of Bone Mineral by Osteocytes by Induction of Carbonic Anhydrase 2. Journal of Bone and Mineral Research 2013, 28, 2436-2448, 10.1002/jbmr.2003.
- Hai Qing; Laleh Ardeshirpour; Paola Divieti Pajevic; Vladimir Dusevich; Katharina Jähn; Shigeaki Kato; John Wysolmerski; Lynda F. Bonewald; Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation.. Journal of Bone and Mineral Research 2012, 27, 1018-29, 10.1002/jbmr.1567.
- Elena Tsourdi; Katharina Jähn; Martina Rauner; Björn Busse; Lynda F. Bonewald; Physiological and pathological osteocytic osteolysis. Journal of musculoskeletal & neuronal interactions 1970, 18, 292-303.
- Wei Wei; Daniel Zeve; Jae Myoung Suh; XueQian Wang; Yang Du; Joseph E. Zerwekh; Paul C. Dechow; Jonathan M. Graff; Yihong Wan; Biphasic and Dosage-Dependent Regulation of Osteoclastogenesis by β-Catenin ▿. Molecular and Cellular Biology 2011, 31, 4706-4719, 10.1128/MCB.05980-11.
- Asiri R. Wijenayaka; Masakazu Kogawa; Hui Peng Lim; Lynda F. Bonewald; David M. Findlay; Gerald J. Atkins; Sclerostin Stimulates Osteocyte Support of Osteoclast Activity by a RANKL-Dependent Pathway. PLOS ONE 2011, 6, e25900, 10.1371/journal.pone.0025900.
- Megan M Weivoda; Ming Ruan; Christine M Hachfeld; Larry Pederson; Alan Howe; Rachel A. Davey; Jeffrey D Zajac; Yasuhiro Kobayashi; Bart O. Williams; Jennifer J Westendorf; et al.Sundeep KhoslaMerry Jo Oursler Wnt Signaling Inhibits Osteoclast Differentiation by Activating Canonical and Noncanonical cAMP/PKA Pathways.. Journal of Bone and Mineral Research 2015, 31, 65-75, 10.1002/jbmr.2599.
- S. L. Holmen; Cassandra R. Zylstra; Aditi Mukherjee; Robert E. Sigler; Mary L. Bouxsein; Lianfu Deng; Thomas L Clemens; M.-C. Faugère; Bart O. Williams; Essential Role of -Catenin in Postnatal Bone Acquisition. Journal of Biological Chemistry 2005, 280, 21162-21168, 10.1074/jbc.m501900200.
- Cecilia M. Giachelli; Ectopic Calcification. The American Journal of Pathology 1999, 154, 671-675, 10.1016/s0002-9440(10)65313-8.
- Cecilia M. Giachelli; Ectopic Calcification. The American Journal of Pathology 1999, 154, 671-675, 10.1016/s0002-9440(10)65313-8.

