 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sushant Khanal | + 326 word(s) | 326 | 2021-03-02 09:41:19 | | | |
| 2 | Vivi Li | + 4025 word(s) | 4351 | 2021-03-10 02:29:22 | | |
Video Upload Options
Eradication of latent human immunodeficiency virus (HIV) infection is a global health challenge. Reactivation of HIV latency and killing of virus-infected cells, the so-called “kick and kill” or “shock and kill” approaches, are a popular strategy for HIV cure. While antiretroviral therapy (ART) halts HIV replication by targeting multiple steps in the HIV life cycle, including viral entry, integration, replication, and production, it cannot get rid of the occult provirus incorporated into the host-cell genome. These latent proviruses are replication-competent and can rebound in cases of ART interruption or cessation. In general, a very small population of cells harbor provirus, serve as reservoirs in ART-controlled HIV subjects, and are capable of expressing little to no HIV RNA or proteins. Beyond the canonical resting memory CD4+ T cells, HIV reservoirs also exist within tissue macrophages, myeloid cells, brain microglial cells, gut epithelial cells, and hematopoietic stem cells (HSCs). Despite a lack of active viral production, latently HIV-infected subjects continue to exhibit aberrant cellular signaling and metabolic dysfunction, leading to minor to major cellular and systemic complications or comorbidities. These include genomic DNA damage; telomere attrition; mitochondrial dysfunction; premature aging; and lymphocytic, cardiac, renal, hepatic, or pulmonary dysfunctions. Therefore, the arcane machineries involved in HIV latency and its reversal warrant further studies to identify the cryptic mechanisms of HIV reservoir formation and clearance.
1. Introduction
HIV latency reversion in many cell types using various reversion agents is discussed in this review. These multiple cell types and different homeostasis in HIV latency are focused. Meanwhile, molecules such as TOX, protein kinases, JAK/STAT, some cell metabolism components (such as apoptotic proteins), as well as some genetic and transcriptional modulators are discussed in different HIV latent models with a focus on either latency establishment, upkeep, or reversion. The involvement of these various cellular components in latency is an important aspect of HIV pathogenesis.
2. HIV Latency and Potential Agents for Reversal
2.1. TOX in HIV Latency
Thymocyte selection-associated high-mobility group box (TOX) is an important player in T cell differentiation due to the presence of an adjacent lysine-rich region that may serve as a nuclear localization signal (NLS), facilitating TOX–DNA interactions and protein–protein interactions. The TOX gene family comprises four major isoforms (TOX1, TOX2, TOX3, and TOX4) in different chromosomal loci. TOX1 regulates immune differentiation, TOX2 regulates natural killer (NK) cells and is a DNA-binding transcription factor that functions in proximity to RNA polymerase II, TOX3 prevents neuronal death, and TOX4 is involved in the process of DNA damage repair and cell-cycle progression from mitosis to interphase [1][2][3][4][5][6][7][8]. Extremely high expression of TOX1 has been observed in microarray analysis of thymic transcripts in CD4+ CD8+ double-positive (DP) thymocytes [1]. This finding indicates the importance of TOX in immune cell differentiation. Furthermore, CD4 T cells, NK cells, and lymph nodes express high levels of TOX1. Failure of T cell and NK cell development and dysfunction of the transcription factor FOXP3 regulated gene expressions are observed in the absence of TOX1, further demonstrating its role in lymphocyte differentiation and functions [3]. Additionally, TOX is also significantly upregulated during tumor generation and progression [4]. A direct interaction between platinated DNA and TOX4 is required for DNA repair in cancer when platinating anticancer drugs, such as cisplatin, trigger DDR [6]. Surprisingly, on the other hand, some reports also suggest that TOX actually inhibits DNA repair by directly binding to KU70/80 and suppressing nonhomologous end joining (NHEJ) repair in T cell acute lymphoblastic leukemia (T-ALL), and when TOX was stably knocked down, it elevated NHEJ repair during DNA double-stranded breaks (DSB). TOX mutants lacking the NLS and high-mobility group (HMG) domain did not alter the NHEJ repair because NLS and HMG directly bind to KU70/80 via HMG domain [9]. These findings suggest the complexity and importance of TOX overall.
A significant decrease in HIV infectivity has been observed during an interplay of TOX4 and its interaction with the PWWP (Pro-Trp-Trp-Pro motif) interacting region (PIR) of a RNA binding protein and a splicing cofactor NOVA1 [10]. The PIR region of NOVA1 and the PIR region of TOX4 in TOX4 HMG domain interact with the PWWP domain of Lens epithelium-derived growth factor (LEDGF/p75), and when all three proteins (TOX4, NOVA1, and LEDGF) are localized in the nucleus and attached to the chromatin, they ultimately and specifically decrease HIV infectivity, but not murine leukemia virus (MLV) infectivity [10]. TOX and its association with high-mobility group box protein 1 (HMGB1) has been shown to play a role in HIV latency maintenance in dendritic cells via crosstalk with NK cells [11]. In epithelial cells, HMGB1 represses HIV replication by inhibiting long terminal repeat (LTR)-mediated transcription of the virion [12]. In contrast, the interaction between HMGB1 and TLR ligands is known to reverse HIV latency in chronically infected U-1 cells in the presence of bacterial components, such as flagellin, bacterial lipopolysaccharides (LPS), and CpG DNA oligos [13]. Importantly, TOX can bind to DNA in a sequence-independent manner to control transcription of a set of genes, such as CCR7, SELL, IL7R, NFAT5, LY6C1, CD38, CTLA4, LAG3, PDCD1, and many other genes associated with T cell longevity and maturation [14][15]. Thus, the dual role of the HMGB1 protein family and the widespread influence of TOX warrants a thorough investigation in order to elucidate their role in HIV latency maintenance and reversal (Figure 2). In a broader sense, TOX is transcriptionally, epigenetically, metabolically, and biochemically involved in T cell longevity, maturity, exhaustion, and survival. So far, there are no reports on specific compounds that can target TOX protein for HIV latency reversal. In silico analysis and molecular docking techniques identified a total of 140 compounds that bind the HMG box domain of the TOX protein, by virtual screening of 7.6 million agents from the ZINC15 database (118 identified) and screening of 200,000 other small molecules (22 identified) [16]. These 140 compounds were tested in vitro in a TOX-dependent Hut7b cell line model of cutaneous T cell lymphoma (CTCL), and 18 molecules were shown to inhibit TOX in both a TOX-high model (Hut78, SZ4, Jurkat cell lines) and a TOX-low model (K562, U937, and Mac2A) [16]. Antibodies and compounds that target HMG box have been reviewed in detail elsewhere, such as Anti-HMGB1 m2G7, acetylcholine, P5779, and resveratrol, which inhibit the signaling pathways associated with HMGB1 and TLR4 signaling [17][18][19]. Moreover, other HMG box protein-binding molecules, such as the receptor for advanced glycation products (RAGE), revealed a link between HIV infection and TLR4 signaling, and the use of RAGE/HMGB1 inhibitors such as FPS-ZM1 in latency study has already been evaluated [20][21][22][23]. Some of these molecules are potential candidates for future studies on HMG box protein and TOX-mediated HIV latency reversion or upkeep. Thus, mechanistic studies of TOX family proteins and their interacting components (ranging from chemicals compounds, antibodies, and RNA–DNA to protein–protein interactions) will help to understand the processes of immune-cell differentiation in long-lived, latently HIV-infected populations.
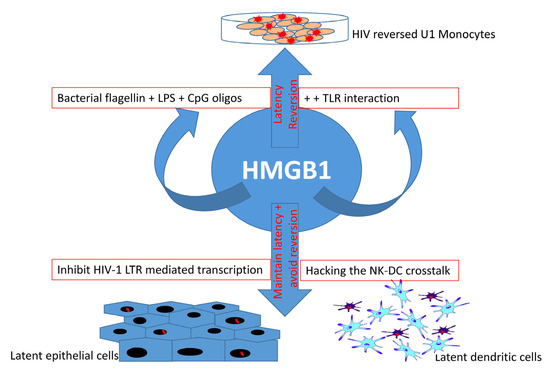
Figure 2. Dual role of HMGB1 in HIV latency. HMGB1 protein not only helps maintain viral latency in epithelial cells and dendritic cells, but also helps reverse HIV latency in U-1 monocytes. This reversal is associated with TLR interaction with bacteria-derived molecules (flagellin, LPS, and CpG oligos) that play a major role in HMGB1-associated reversal, suggesting the importance of foreign (bacterial) components in HIV reversal. However, as an indication of a dual role of HMGB1 protein family, latency is maintained and its reversion is prevented through the inhibition of HIV-1 LTR-mediated transcription and hacking the natural killer (NK) cells and dendritic cell (DC) crosstalk in latent epithelial cells and dendritic cells, respectively.
2.2. Protein Kinases in HIV Latency
Protein kinases and their roles in HIV latency/reservoir maintenance/reversal have been described previously [24][25]. A recent study using 418 structurally diverse, cell-permeable, and medicinally active kinase inhibitors in a latent cell line model showed that control of kinase activity can affect a wide range of cellular pathways or signaling cascades and block HIV-1 latency reversal [26]. One such multikinase inhibitor, midostaurin, plays a dual role by activating latency reversal and blocking viral replication or reversal in the presence or absence of SAM domain and HD domain-containing protein 1 (SAMDH1), respectively [27][28]. Kinome profiling recognizes the contribution of PIM-1 kinase in latent HIV infection and reactivation in T cell lines, as well as in primary CD4 T cells [29]. In both models, HIV reactivation is largely affected following inhibition or knockdown of PIM-1 by the PIM-1 inhibitor IV (PIMi IV) or PIM-1 specific shRNAs, respectively [29]. Derivatives of the compound benzolactam promote apoptosis in ACH-2 and J-lat cells via a protein kinase C (PKC)-induced latency-reversal pathway, and one such derivative (BL-V8-310) was found to have high LRA activity that also reduced cytotoxic cytokine secretion [30]. These studies reveal the prevailing importance of protein kinases in HIV-1 reservoir/latency maintenance and/or reversal. The individual kinase signaling pathways that are affected during HIV latency reversal have been described previously [26].
The mammalian target of rapamycin (mTOR) protein is a serine/threonine protein kinase that belongs to the PI3K-related kinase family and is associated with a wide range of cellular processes such as cell proliferation, motility, growth, and survival, as well as protein synthesis and gene transcription. The interactions between dendritic cells and T cells through cell surface receptors/ligands, which activate PI3K-Akt-mTOR signaling by triggering dephosphorylation of proteins downstream of the Akt signaling pathway [31], play an important role in HIV latency reversal. However, such activation does not increase the expression/activity of nuclear transcription factors, such as NF-κB. Even blockade of c-Fos and c-Jun transcription factors, which directly bind to HIV-1 LTR promoter regions, does not affect dendritic cell contact-mediated latency reversal [31]. Also, HIV Tat drives latent provirus to an active virus-producing state by recruiting pTEFb complex and interacting with the viral mRNA hairpin in the HIV promoter region [32]. Cyclin T1 and CDK9 form the pTEFb protein complex to act with Tat as a cofactor for Tat-mediated transcription [33]. The CDK9 partner, cyclin T1, specifically enhances binding of Tat protein in the trans-activation response element (TAR) RNA stem loop structure [34][35][36]. The 42 kDa kinase CDK9, when mutated/inhibited, blocks Tat transactivation without exhibiting any effects in the overall T cell activation, which proves that CDK9 kinase activity is essential for Tat activation, which subsequently affects pTEFb recruitment and TAR RNA association [36][37][38][39][40][41]. Moreover, the amount of CDK9 phosphorylation and kinase activity can have direct effects on Tat association, and the degree of latency establishment; also, site-directed mutations within different locations within Tat protein residues revealed the importance of certain sites in the CDK9-Tat interplay in a crystallized model and in silico analysis of Tat-CDK9-Cyclin T complex [42][43]. The Tat inhibitor didehydro-Cortistatin A (dCA) can prevent the HIV transcription by blocking the assembly of Tat in HIV promoter with specific transcription factors and RNA polymerase II (RNAPII)-associated proteins, preventing the assembly of the transcription initiation complex, which eventually drives the cell toward a latent state (Figure 3) [32]. However, suppressing the multifunctional mTOR protein activity using mTOR inhibitors minimizes latency reversal in both a Tat-dependent and a Tat-independent manner by blocking CDK9 phosphorylation [33], stressing the importance of mTOR in latency. Another transcriptional regulatory factor, KAP1, is critical for the reactivation of HIV latency, which recruits CDK9 and interacts with the proviral promoter region allowing viral transcription [44]. Surprisingly, HIV-host transcription mechanisms have developed a bypass machinery to avoid KAP1-mediated activation in order to reduce the magnitude of virus production and latency reactivation [44]. Investigating the interplay between CDK9, KAP1, mTOR, and Tat may enhance our understanding of the HIV latency.
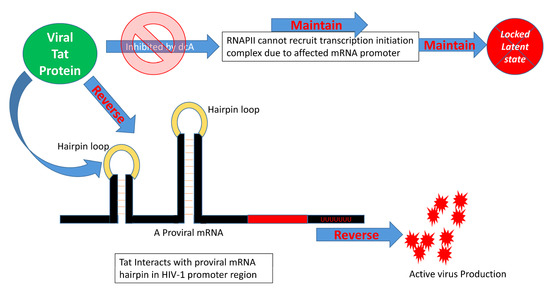
Figure 3. HIV Tat protein and latency reversal. Tat protein can reverse HIV latency by interacting with the promoter region of the HIV genome through recruiting RNAPII and associated transcription initiation complex. This process is affected by didehydro-cortistatin A (dCA). When RNAPII becomes unable to interact with the HIV mRNA hairpin loop, the latency reversal is halted. A thorough investigation of the roles of individual viral proteins in HIV latency reversal is thus very important for understanding their critical role in HIV cure.
2.3. JAK/STAT Pathway in HIV Latency
A widely studied kinase cascade—the Janus kinase (JAK) signal transducer and activator of transcription (STAT) pathway (Figure 1)—has an important role in immune-cell activation, polarization, cytokine signaling, innate and adaptive immune responses, and autoimmune disease development [45][46][47][48]. The function of JAK/STAT in maintaining CD4 T cell progression has been reviewed previously [45]. Since latent reservoirs largely comprise CD4+ helper T cells, which have dual roles in acute and chronic HIV infection, investigating the role of the JAK/STAT pathway in establishing and maintaining the HIV reservoir in helper T cells is essential for increasing the understanding of their reversal [49][50][51][45][46][47]. The activation of JAK/STAT and the central regulator mTOR pathways inhibit HIV reactivation and prevent the production of virus particles from a latent state. However, inhibition of the JAK/STAT pathway promotes HIV-1 reversion [52]. In contrast, when JAK is inhibited using JAK inhibitors, such as tofacitinib and ruxolitinib, inhibition of viral production is observed in HIV reservoirs via suppression of an IL-15-mediated viral reactivation pathway [53][54]. These studies highlight the dual and complex role of the JAK/STAT pathway in HIV latency and reversal (Figure 1), and present this molecular pathway as a mechanism that warrants further investigation. STAT plays an important role in HIV development independent of the infection status (acute or chronic), and a use of benzotriazoles reactivates HIV latency by preventing a negative feedback loop carried out by SUMO2/3 (affecting phosphorylated STAT5), which sustained the STAT5 phosphorylation and its active form [55][56][57]. In addition, HIV-1 expression was restricted in U-1 latent cell lines following a heterodimer complex formation between p50 (viral promoter binding protein) and naturally occurring C-terminally truncated STAT5 [58]. The availability of STAT in its active form is important for latency reversion. On the other hand, STAT3 inhibitor 5,15-DPP at 50 and 500 nM has been shown to promote HIV-1 transmission and reversion when compared to 5 nM and no treatment using an envelope defective mutant HIV strain [52][59]. However, the STAT1 inhibitor fludarabine was shown to block IL-6 and HIV-1 interplay, reducing the monocyte migration and damage in a recent study [60]. Phosphorylation of STAT 1, 3, and 5 by IFN-α, but not others (IFN-β, ω, ε, λ1, and λ3) was successful in reversing HIV latency using in vitro cell models, as well as in CD4 T cells derived from patients undergoing ART [61]. The study of specific viral components in relation to the JAK/STAT pathway is essential to identify new targets for HIV latency reversal. For example, the viral accessory protein Vif is directly involved in the degradation of the JAK/STAT pathway [62]. Vif interacts with STAT1 and STAT3, but not STAT2, and plays a critical role in the prevention of the antiviral effects of Type-1 IFN-α signaling [62]. Therefore, the roles and mechanistic effects of LRAs, protein interactions, IFN activities, and cytokine signaling on the JAK/STAT pathway and on viral proteins should be investigated in detail in order to understand their potentials as molecular targets in HIV latency maintenance and/or reversal.
2.4. Apoptotic Proteins in HIV Latency
Productive viral infection, latency establishment, and latency reversion affect cellular metabolism. Cellular metabolism is hijacked by HIV-1 and changes in antioxidation, iron metabolism, and iron import are observed during latency transitions, while oxidative stress is increased and antioxidant response is upregulated during latency reversion and also during productive in vitro and in vivo infections [63]. Drugs increasing oxidative stress or iron content and an increase in antioxidant gene expression resulted in reactivation of latency, causing degradation of promyelocytic leukemia protein nuclear bodies [63]. Another drug, auranofin, which is used to treat rheumatoid arthritis, had an impact on the latent viral reservoir by reducing the number of the integrated viral DNA in HIV patients under ART [64]. Partial inhibition of glycolysis using 2-deoxy glucose blocked HIV-1 infection, decreased cell viability of preinfected cells, and most importantly, avoided latency reversion in CD4 T cells obtained from HIV patients under ART [65]. Uninfected and HIV-infected macrophages use fatty acid and glucose as primary sources of energy, however, latent macrophages used glutamine/glutamate as a major source of energy, and blocking the glutamine, glutamate, and alpha-ketoglutarate pathways killed latent HIV-infected macrophages [66]. Viral infections alter, hack, and reprogram host metabolism regularly, as discussed before [67][68]. Thus, there is a strong connection between latency upkeep/reversion and metabolic pathways, agents, drugs, etc.
Viral proteins such as Nef, Tat, Vpu, GP120, and Vpr have been shown to promote or inhibit cell apoptosis [69]. Many of the differential regulatory effects of these proteins are dependent upon the phase of HIV in infected cells. For instance, many of these proteins are anti-apoptotic during acute infection to enable persistent infection, but may transition to pro-apoptotic in the process of establishing chronic infection by inducing bystander apoptosis. The role of the envelope and protease proteins in activating apoptosis has been reviewed elsewhere [70][69]. Intriguingly, some viral proteins can have dual roles, with both pro- and anti-apoptotic effects [70][69]. Downregulation of pro-apoptotic proteins and/or upregulation of anti-apoptotic proteins are key to escaping apoptosis, and thus favor survival of latent cells. For example, Debio 1143 is an inhibitor of anti-apoptotic proteins that activates HIV transcription via NF-κB signaling by degrading BIRC2 protein that mediates anti-apoptotic effects on latently HIV-infected cells [71]. Figure 4 illustrates how Debio 1143 can reverse HIV-1 latency in resting CD4 T cells derived from peripheral blood mononuclear cells (PBMCs) of HIV patients and humanized BLT mice on ART treatment [71]. On the other hand, Debio 1143, in combination with an anti-PD1 mAb, exhibits a significant PD-1 blockade-mediated HIV reduction in all tissues (spleen, lymph nodes, liver, lungs, and thymic organoids) in BLT mice [72]. Enhanced expression of the anti-apoptotic protein BIRC5 and its upstream regulator OX40 in productively and latently-infected CD4 T cells promotes HIV-infected cell survival [73], whereas inhibition of these molecules enhances HIV-infected cell death [73].
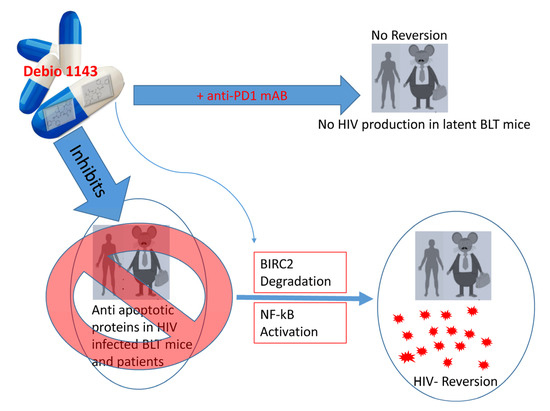
Figure 4. Inhibition of anti-apoptotic proteins reverses HIV latency. Debio 1143 (an IAP inhibitor) inhibits anti-apoptotic proteins such as BIRC2, which activates NF-κB-mediated HIV reversal. HIV latency can be markedly reversed in ART-treated BLT mice and human subjects when treated with Debio 1143. On the other hand, Debio 1143, when combined with anti-PD1 monoclonal antibody (mAb), can reduce HIV production and latency reversion in BLT humanized mice in most tissues (spleen, lymph nodes, liver, lungs, and thymic organoids). The dual role of chemical agents in HIV reversal/maintenance is thus intriguing and warrants further investigation.
The tumor suppressor protein p53 can eliminate HIV-infected cells by enhancing the expression of PTEN—a negative regulator of protein kinase B/AKT [70][74][75]. AKT couples with PIP-3 of the PIK3 signaling pathway and activates itself, which inhibits pro-apoptotic proteins while also coactivating anti-apoptotic proteins, such as pBad, Bcl-2, and FOX01 transcription factor [70][76][77]. Triggering receptor expressed on myeloid cells 1 (TREM1) silencing in HIV-infected macrophages reduces anti-apoptotic protein expression, increases pro-apoptotic signaling, and affects the survival of HIV-infected cells, leading to disruption of the mitochondrial membrane potential, cytochrome-C release, and caspase-9 cleavage [78]. HIV promotes latent cell survival in a TREM1-dependent fashion and manipulates the anti-apoptotic Bcl-2 protein family [78]. Thus, these signaling components can promote latent cell survival and HIV reservoir establishment. Interactions between HIV-1 and the Bcl-2 protein family, as well as possible therapeutics, have been reviewed elsewhere [79]. A detailed investigation of these pro- and anti-apoptotic proteins in latency establishment and maintenance is essential to develop novel reversal approaches [70]. New studies on the association between the apoptosis-related proteins and HIV latency may lead to better approaches to controlling their expression levels or developing new drugs to regulate their expression, either individually or in combination (e.g., Debio 1143 in combination with anti-PD1 mAb) (Figure 4) [72]. Additionally, proteins associated with cell-death mechanisms other than apoptosis during viral infection, such as autophagy (LC3B, SQSTM1/p62), pyroptosis (Caspase1, 3), ferroptosis (GPX4), and necroptosis (RIP1, 3), should be investigated in order to understand their roles in HIV latency maintenance and/or reversal [80][81][82][83][84][85][86][87][88].
2.5. Transcriptional and Genetic Factors in HIV Latency
In the CCL19 and IL-7 treated resting CD4 T latency model described above, an increase in the levels of three microRNAs (miRs) such as miR98, miR4516, and miR7974, was shown by next-generation sequencing, however inhibiting these miRNAs did not reverse latency [89]. It is clear that inhibiting these miRNAs might not only reverse latency, and more mechanisms/aspects are involved in it. However, the fact that these miRNAs were upregulated in these latent HIV models should not be ignored, and thus further studies of these noncoding genetic elements and other potential regulators seem to be essential for understanding the mechanisms of HIV latency. Another miRNA known as TAR was abundant in exosomal vesicles in supernatants collected from an in vitro infection model, as well as serum, cerebrospinal fluid, blood plasma and even in saliva from patients under ART and in serum from HIV-1 infected humanized mice [90][91][92][93]. During HIV latency, the incorporated proviral genome is extremely low (only 1-5 copies), but TAR RNA copy numbers range from 103 to 105 in patients undergoing ART suggesting that a true/complete transcriptional latency is not always the case [91][94]. As described previously, TAR RNA is essential for HIV Tat and protein kinase(s) RNA-activated (PKR) binding, however, the TAR miRNA binds to TLR7/TLR8, but not PKR [91]. Coculturing exosomal vesicles containing Tat, TAR RNA, TAR miRNA, and a newly found TAR-gag RNA induced IL-6, TNFβ, NF-κB pathways, cytokine production, and overall cellular activation of the recipient/neighbor cells, ultimately causing latency reversion [91][92][94][95]. These TAR RNA-containing exosomes also induced cancer-cell proliferation and progression by affecting expression of proto-oncogenes and TLR3 inducible genes, which reduced apoptosis in neighboring latency-reversing cells by lowering Bim and CDK9 protein levels [90][96]. These exosomal vesicles also contain phosphorylated c-Src, which causes PI3K-mTOR-AKT-mediated and P300/SRC-1, STAT3-activated latency reversion [95]. In addition, high levels of NF-κB and P300 were observed in the nuclei of latent cells cocultured with exosomal vesicles [95]. These vesicles can be targeted with antibodies, antibiotics, drugs, transcription inhibitors, and ARTs to alter the ratio/amount of their individual components and subsequently alter their roles in HIV latency upkeep/reversion [97][98]. For example, ARTs (Indinacir and Emitricitabine), antibiotics (oxytetracycline, tetracycline, methacycline, and demeclocycline), and even interferon treatments showed effects on the proteins involved in the endosomal sorting complex required for transport (ESCRT) pathway and caused changes in TAR RNA levels and other contents of these exosomal vesicles [97]. In addition, incorporation of a transcriptional inhibitor F07#13 in a mathematical model in various cell types displayed potential to induce changes in HIV latency and LTR dynamics and were different among the cell types used [98]. In a study that used a library of FDA-approved drugs, HIV-1 proviral transcription was activated with febuxostat, eltrombopag, and resveratrol, while mycophenolate inhibited HIV-1 proviral transcription, and these transcriptional modulators exhibited different effects in different cell types (lymphoid versus myeloid lineage) [99].
A newly identified QUECEL (quiescent effector cell latency) model used polarized T cell subsets (Th1, Th2, Th17, and TREG) to mimic HIV latency in vitro [100]. This model represents the escape of latently infected cells from cell-cycle checkpoints and showed a significant reduction in cell-cycle-dependent cyclins D3 and B1, and restricted expression of cell-activation markers such as CD69 and CD25, and the positive transcription elongation factor P-TEFb (Figure 1) [100]. RNA-Seq and follow-up studies in quiescent cells suggests that altered gene expression is associated with latent cell expansion, reinforcing the roles of cyclin-dependent kinase pTEFb induction and CDK9 phosphorylation in latency reactivation and the anti-inflammatory molecule TGF-β elevation in latency maintenance [100][101][102]. Notably, the QUECEL model revealed alterations in regulatory machineries, indicating that the c-Myc pathways are highly repressed and NF-κB is largely dispensable, whereas nuclear factor of activated T cells (NFAT) and NFAT-dependent latency reactivation are required for HIV latency [100]. NF-κB and NFAT binding at sites in the proviral enhancer are calcium-dependent and positively regulate viral transcription as well as T cell activation, and the NF-κB, NFAT, when bound can recruit a histone acetyltransferase, p65, in LTR regions to promote HIV transcription [103][104][105][106][107][108].
The HIV genome promoter regions U3 and R, which have a greater involvement of the 5′-long terminal repeat (LTR) section and a minimal participation of the 3′-LTR section, are responsible for controlling viral expression following different mechanisms of transcriptional interference in these regions of the viral genome, such as promoter occlusion and steric hindrance, because the viral DNA blocks the active and normal transcriptional process within the region of its incorporation in the host-cell chromosome [109][110][111][112][113]. The 5′-LTR region is very important in controlling transcription because the interaction between negative elongation factor (NELF) and RNAPII can lead to premature termination of transcription to limit the escape of the transcriptional complexes [109][110][111][112][113][114][115]. The mechanisms involved in controlling histone deacetylation and methylation, along with the use of long noncoding RNAs (lncRNAs) to repress the latency reversal, has been reviewed elsewhere [109]. Previous studies showed that a single nucleotide polymorphism (SNP) in a genomic region with close proximity to the CCR5 coding region could control HIV-1 associated coreceptor CCR5 mRNA expression via CCR5AS-lncRNA-mediated sequestering of Raly, a protein that binds and degrades CCR5 mRNA [116]. The reduction in CCR5-lncRNA failed to protect the CCR5-mRNA and resulted in a very low expression of CCR5 on the cell surface [116]. The ability to manipulate the expressions of these coreceptors during acute/chronic stages of viral infection could have an effect on HIV latency. Another SNP (rs2027820) with a virtually perfect linkage disequilibrium with the previous (rs1015164) SNP also controls the coreceptor CCR5 through differential binding of activating transcription factor (ATF1) [116]. These studies imply that future research should focus on such noncoding elements, polymorphic areas, SNPs, etc. The importance of the genomic noncoding regions in HIV pathogenesis could be a new area of investigation, which should focus on the role of these noncoding RNA sequences during latency initiation, maintenance/upkeep, and reversal [116].
Notably, the mechanisms associated with the host mRNA decay and the proteins involved in active HIV infection are quite different from those involved in latent HIV infection. The host mRNA decay proteins UPF1, UPF2, SMG6, and Staufen1 were significantly downregulated in monocyte-derived macrophages (MDMs) infected with HIV [117]. UPF2 and SMG6 downregulation via siRNA-mediated silencing enhances HIV gene expression; however, Staufen1 silencing impairs HIV gene expression [117]. Further investigation of the viral proteins involved in these cellular defense machineries is warranted and could uncover more molecular and genetic targets needed for HIV cure.
References
- Wilkinson, B.; Chen, J.Y.-F.; Han, P.; Rufner, K.M.; Goularte, O.D.; Kaye, J. TOX: An HMG box protein implicated in the regulation of thymocyte selection. Nat. Immunol. 2002, 3, 272–280.
- Yun, S.; Lee, S.H.; Yoon, S.-R.; Kim, M.S.; Piao, Z.-H.; Myung, P.-K.; Kim, T.-D.; Jung, H.; Choi, I. TOX regulates the differentiation of human natural killer cells from hematopoietic stem cells in vitro. Immunol. Lett. 2011, 136, 29–36.
- Aliahmad, P.; Kaye, J. Development of all CD4 T lineages requires nuclear factor TOX. J. Exp. Med. 2008, 205, 245–256.
- Yu, X.; Li, Z. TOX gene: A novel target for human cancer gene therapy. Am. J. Cancer Res. 2015, 5, 3516–3524.
- Dittmer, S.; Kovacs, Z.; Yuan, S.H.; Siszler, G.; Kögl, M.; Summer, H.; Geerts, A.; Golz, S.; Shioda, T.; Methner, A. TOX3 is a neuronal survival factor that induces transcription depending on the presence of CITED1 or phosphorylated CREB in the transcriptionally active complex. J. Cell Sci. 2010, 124, 252–260.
- Du Puch, C.B.M.; Barbier, E.; Kraut, A.; Coute, Y.; Fuchs, J.; Buhot, A.; Livache, T.; Sève, M.; Favier, A.; Douki, T.; et al. TOX4 and its binding partners recognize DNA adducts generated by platinum anticancer drugs. Arch. Biochem. Biophys. 2011, 507, 296–303.
- Vong, Q.P.; Leung, W.-H.; Houston, J.; Li, Y.; Rooney, B.; Holladay, M.; Oostendorp, R.A.J.; Leung, W. TOX2 regulates human natural killer cell development by controlling T-BET expression. Blood 2014, 124, 3905–3913.
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415.
- Lobbardi, R.; Pinder, J.; Martinez-Pastor, B.; Theodorou, M.; Blackburn, J.S.; Abraham, B.J.; Namiki, Y.; Mansour, M.; Abdelfattah, N.S.; Molodtsov, A.; et al. TOX Regulates Growth, DNA Repair, and Genomic Instability in T-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2017, 7, 1336–1353.
- Morchikh, M.; Naughtin, M.; Di Nunzio, F.; Xavier, J.; Charneau, P.; Jacob, Y.; Lavigne, M. TOX4 and NOVA1 Proteins Are Partners of the LEDGF PWWP Domain and Affect HIV-1 Replication. PLoS ONE 2013, 8, e81217.
- Gougeon, M.-L.; Melki, M.-T.; Saïdi, H. HMGB1, an alarmin promoting HIV dissemination and latency in dendritic cells. Cell Death Differ. 2011, 19, 96–106.
- Naghavi, M.H.; Nowak, P.; Andersson, J.; Sönnerborg, A.; Yang, H.; Tracey, K.J.; Vahlne, A. Intracellular high mobility group B1 protein (HMGB1) represses HIV-1 LTR-directed transcription in a promoter- and cell-specific manner. Virology 2003, 314, 179–189.
- Nowak, P.; Abdurahman, S.; Lindkvist, A.; Troseid, M.; Sönnerborg, A. Impact of HMGB1/TLR Ligand Complexes on HIV-1 Replication: Possible Role for Flagellin during HIV-1 Infection. Int. J. Microbiol. 2012, 2012, 1–10.
- O’Flaherty, E.; Kaye, J. TOX defines a conserved subfamily of HMG-box proteins. BMC Genom. 2003, 4, 13.
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218.
- Agrawal, V.; Su, M.; Huang, Y.; Hsing, M.; Cherkasov, A.; Zhou, Y. Computer-Aided Discovery of Small Molecule Inhibitors of Thymocyte Selection-Associated High Mobility Group Box Protein (TOX) as Potential Therapeutics for Cutaneous T-Cell Lymphomas. Molecules 2019, 24, 3459.
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB. Front. Immunol. 2020, 11, 484.
- Yang, H.; Wang, H.; Ju, Z.; Ragab, A.A.; Lundbäck, P.; Long, W.; Valdés-Ferrer, S.I.; He, M.; Pribis, J.P.; Li, J.; et al. MD-2 is required for disulfide HMGB1&ndash, dependent TLR4 signaling. J. Exp. Med. 2015, 212, 5–14.
- Yang, Y.; Li, S.; Yang, Q.; Shi, Y.; Zheng, M.; Liu, Y.; Chen, F.; Song, G.; Xu, H.; Wan, T.; et al. Resveratrol Reduces the Proinflammatory Effects and Lipopolysaccharide- Induced Expression of HMGB1 and TLR4 in RAW264.7 Cells. Cell. Physiol. Biochem. 2014, 33, 1283–1292.
- Vanpatten, S.; Al-Abed, Y. High Mobility Group Box-1 (HMGb1): Current Wisdom and Advancement as a Potential Drug Target. J. Med. Chem. 2018, 61, 5093–5107.
- András, I.E.; Garcia-Contreras, M.; Yanick, C.; Perez, P.; Sewell, B.; Durand, L.; Toborek, M. Extracellular vesicle-mediated amyloid transfer to neural progenitor cells: Implications for RAGE and HIV infection. Mol. Brain 2020, 13, 1–17.
- Liu, L.; Yu, J.; Li, L.; Zhang, B.; Liu, L.; Wu, C.-H.; Jong, A.; Mao, D.-A.; Huang, S.-H. Alpha7 nicotinic acetylcholine receptor is required for amyloid pathology in brain endothelial cells induced by Glycoprotein 120, methamphetamine and nicotine. Sci. Rep. 2017, 7, 40467.
- Yuan, S.; Liu, Z.; Xu, Z.; Liu, J.; Zhang, J. High mobility group box 1 (HMGB1): A pivotal regulator of hematopoietic malignancies. J. Hematol. Oncol. 2020, 13, 1–19.
- Sadowski, I.; Hashemi, F.B. Strategies to eradicate HIV from infected patients: Elimination of latent provirus reservoirs. Cell. Mol. Life Sci. 2019, 76, 3583–3600.
- Stoszko, M.; Ne, E.; Abner, E.; Mahmoudi, T. A broad drug arsenal to attack a strenuous latent HIV reservoir. Curr. Opin. Virol. 2019, 38, 37–53.
- Vargas, B.; Giacobbi, N.S.; Sanyal, A.; Venkatachari, N.J.; Han, F.; Gupta, P.; Sluis-Cremer, N. Inhibitors of Signaling Pathways That Block Reversal of HIV-1 Latency. Antimicrob. Agents Chemother. 2018, 63, e01744-18.
- Garcia-Vidal, E.; Badia, R.; Pujantell, M.; Castellví, M.; Felip, E.; Clotet, B.; Riveira-Muñoz, E.; Ballana, E.; Esté, J.A. Dual effect of the broad spectrum kinase inhibitor midostaurin in acute and latent HIV-1 infection. Antivir. Res. 2019, 168, 18–27.
- Chougui, G.; Margottin-Goguet, F. HUSH, a Link Between Intrinsic Immunity and HIV Latency. Front. Microbiol. 2019, 10, 224.
- Duverger, A.; Wolschendorf, F.; Anderson, J.C.; Wagner, F.; Bosque, A.; Shishido, T.; Jones, J.; Planelles, V.; Willey, C.; Cron, R.Q.; et al. Kinase Control of Latent HIV-1 Infection: PIM-1 Kinase as a Major Contributor to HIV-1 Reactivation. J. Virol. 2013, 88, 364–376.
- Matsuda, K.; Kobayakawa, T.; Tsuchiya, K.; Hattori, S.-I.H.; Nomura, W.; Gatanaga, H.; Yoshimura, K.; Oka, S.; Endo, Y.; Tamamura, H.; et al. Benzolactam-related compounds promote apoptosis of HIV-infected human cells via protein kinase C–induced HIV latency reversal. J. Biol. Chem. 2019, 294, 116–129.
- Van Montfort, T.; Van Der Sluis, R.; Darcis, G.; Beaty, D.; Groen, K.; Pasternak, A.O.; Pollakis, G.; Vink, M.; Westerhout, E.M.; Hamdi, M.; et al. Dendritic cells potently purge latent HIV-1 beyond TCR-stimulation, activating the PI3K-Akt-mTOR pathway. EBioMedicine 2019, 42, 97–108.
- Li, C.; Mousseau, G.; Valente, S.T. Tat inhibition by didehydro-Cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat. Epigenetics Chromatin 2019, 12, 1–17.
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe 2016, 20, 785–797.
- Kulinski, T.; Olejniczak, M.; Huthoff, H.; Bielecki, L.; Pachulska-Wieczorek, K.; Das, A.T.; Berkhout, B.; Adamiak, R.W. The Apical Loop of the HIV-1 TAR RNA Hairpin Is Stabilized by a Cross-loop Base Pair. J. Biol. Chem. 2003, 278, 38892–38901.
- Lu, J.; Kadakkuzha, B.M.; Zhao, L.; Fan, M.; Qi, X.; Xia, T. Dynamic Ensemble View of the Conformational Landscape of HIV-1 TAR RNA and Allosteric Recognition. Biochemistry 2011, 50, 5042–5057.
- Wei, P.; Garber, E.M.; Fang, S.-M.; Fischer, W.H.; Jones, A.K. A Novel CDK9-Associated C-Type Cyclin Interacts Directly with HIV-1 Tat and Mediates Its High-Affinity, Loop-Specific Binding to TAR RNA. Cell 1998, 92, 451–462.
- Garber, M.E.; Mayall, T.P.; Suess, E.M.; Meisenhelder, J.; Thompson, N.E.; Jones, K.A. CDK9 Autophosphorylation Regulates High-Affinity Binding of the Human Immunodeficiency Virus Type 1 Tat–P-TEFb Complex to TAR RNA. Mol. Cell. Biol. 2000, 20, 6958–6969.
- Salerno, D.; Hasham, M.G.; Marshall, R.; Garriga, J.; Tsygankov, A.Y.; Graña, X. Direct inhibition of CDK9 blocks HIV-1 replication without preventing T-cell activation in primary human peripheral blood lymphocytes. Gene 2007, 405, 65–78.
- Mancebo, H.S.; Lee, G.; Flygare, J.; Tomassini, J.; Luu, P.; Zhu, Y.; Peng, J.; Blau, C.; Hazuda, D.; Price, D.; et al. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 1997, 11, 2633–2644.
- Yang, X.; Herrmann, C.H.; Rice, A.P. The human immunodeficiency virus Tat proteins specifically associate with TAK in vivo and require the carboxyl-terminal domain of RNA polymerase II for function. J. Virol. 1996, 70, 4576–4584.
- Zhu, Y.; Pe’Ery, T.; Peng, J.; Ramanathan, Y.; Marshall, N.; Marshall, T.; Amendt, B.; Mathews, M.B.; Price, D.H. Transcription elongation factor P-TEFb is required for HIV-1 Tat transactivation in vitro. Genes Dev. 1997, 11, 2622–2632.
- Budhiraja, S.; Famiglietti, M.; Bosque, A.; Planelles, V.; Rice, A.P. Cyclin T1 and CDK9 T-Loop Phosphorylation Are Downregulated during Establishment of HIV-1 Latency in Primary Resting Memory CD4+ T Cells. J. Virol. 2013, 87, 1211–1220.
- Wang, H.; Song, L.; Zhou, T.; Zeng, C.; Jia, Y.; Zhao, Y. A computational study of Tat–CDK9–Cyclin binding dynamics and its implication in transcription-dependent HIV latency. Phys. Chem. Chem. Phys. 2020, 22, 25474–25482.
- Morton, E.L.; Forst, C.V.; Zheng, Y.; DePaula-Silva, A.B.; Ramirez, N.-G.P.; Planelles, V.; D’Orso, I. Transcriptional Circuit Fragility Influences HIV Proviral Fate. Cell Rep. 2019, 27, 154–171.e9.
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 1–13.
- O’Shea, J.J.; Plenge, R. JAK and STAT Signaling Molecules in Immunoregulation and Immune-Mediated Disease. Immunity 2012, 36, 542–550.
- Rose, N.R. Autoimmune Diseases. Int. Encycl. Public Health 2017, 12, 192–195.
- Darnell, J.E.D., Jr. STATs and Gene Regulation. Science 1997, 277, 1630–1635.
- Perreau, M.; Savoye, A.-L.; De Crignis, E.; Corpataux, J.-M.; Cubas, R.; Haddad, E.K.; De Leval, L.; Graziosi, C.; Pantaleo, G. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J. Exp. Med. 2013, 210, 143–156.
- Banga, R.; Procopio, F.A.; Noto, A.; Pollakis, G.; Cavassini, M.; Ohmiti, K.; Corpataux, J.-M.; De Leval, L.; Pantaleo, G.; Perreau, M. PD-1+ and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat. Med. 2016, 22, 754–761.
- Noto, A.; Procopio, F.A.; Banga, R.; Suffiotti, M.; Corpataux, J.M.; Cavassini, M.; Riva, A.; Fenwick, C.; Gottardo, R.; Perreau, M.; et al. CD32+ and PD-1+ lymph node CD4 T cells support persistent HIV-1 transcription in treated aviremic individuals. J. Virol. 2018, 92, e00901-18.
- Quan, Y.; Xu, H.; Han, Y.; Mesplède, T.; Wainberg, M.A. JAK-STAT Signaling Pathways and Inhibitors Affect Reversion of Envelope-Mutated HIV. J. Virol. 2017, 91, e00075-17.
- Gavegnano, C.; Brehm, J.H.; Dupuy, F.P.; Talla, A.; Ribeiro, S.P.; Kulpa, D.A.; Cameron, C.; Santos, S.; Hurwitz, S.J.; Marconi, V.C.; et al. Novel mechanisms to inhibit HIV reservoir seeding using Jak inhibitors. PLoS Pathog. 2017, 13, e1006740.
- Gavegnano, C.; Detorio, M.; Montero, C.; Bosque, A.; Planelles, V.; Schinazi, R.F. Ruxolitinib and Tofacitinib Are Potent and Selective Inhibitors of HIV-1 Replication and Virus Reactivation In Vitro. Antimicrob. Agents Chemother. 2014, 58, 1977–1986.
- Venkatachari, N.J.; Zerbato, J.M.; Jain, S.; Mancini, A.E.; Chattopadhyay, A.; Sluis-Cremer, N.; Bar-Joseph, Z.; Ayyavoo, V. Temporal transcriptional response to latency reversing agents identifies specific factors regulating HIV-1 viral transcriptional switch. Retrovirology 2015, 12, 1–22.
- Bosque, A.; Nilson, K.A.; Macedo, A.B.; Spivak, A.M.; Archin, N.M.; Van Wagoner, R.M.; Martins, L.J.; Novis, C.L.; Szaniawski, M.A.; Ireland, C.M.; et al. Benzotriazoles Reactivate Latent HIV-1 through Inactivation of STAT5 SUMOylation. Cell Rep. 2017, 18, 1324–1334.
- Selliah, N.; Zhang, M.; DeSimone, D.; Kim, H.; Brunner, M.; Ittenbach, R.F.; Rui, H.; Cron, R.Q.; Finkel, T.H. The γc-cytokine regulated transcription factor, STAT5, increases HIV-1 production in primary CD4 T cells. Virology 2006, 344, 283–291.
- Della Chiara, G.; Crotti, A.; Liboi, E.; Giacca, M.; Poli, G.; Lusic, M. Negative Regulation of HIV-1 Transcription by a Heterodimeric NF-κB1/p50 and C-Terminally Truncated STAT5 Complex. J. Mol. Biol. 2011, 410, 933–943.
- Quan, Y.; Xu, H.; Kramer, V.G.; Han, Y.; Sloan, R.D.; Wainberg, M.A. Identification of an env-defective HIV-1 mutant capable of spontaneous reversion to a wild-type phenotype in certain T-cell lines. Virol. J. 2014, 11, 177.
- Chaudhuri, A.; Yang, B.; Gendelman, H.E.; Persidsky, Y.; Kanmogne, G.D. STAT1 signaling modulates HIV-1–induced inflammatory responses and leukocyte transmigration across the blood-brain barrier. Blood 2008, 111, 2062–2072.
- Van Der Sluis, R.M.; Zerbato, J.M.; Rhodes, J.W.; Pascoe, R.D.; Solomon, A.; Kumar, N.A.; Dantanarayana, A.I.; Tennakoon, S.; Dufloo, J.; McMahon, J.; et al. Diverse effects of interferon alpha on the establishment and reversal of HIV latency. PLoS Pathog. 2020, 16, e1008151.
- Gargan, S.; Ahmed, S.; Mahony, R.; Bannan, C.; Napoletano, S.; O’Farrelly, C.; Borrow, P.; Bergin, C.; Stevenson, N.J. HIV-1 Promotes the Degradation of Components of the Type 1 IFN JAK/STAT Pathway and Blocks Anti-viral ISG Induction. EBioMedicine 2018, 30, 203–216.
- Shytaj, I.L.; Lucic, B.; Forcato, M.; Penzo, C.; Billingsley, J.; Laketa, V.; Bosinger, S.; Stanic, M.; Gregoretti, F.; Antonelli, L.; et al. Alterations of redox and iron metabolism accompany the development of HIV latency. EMBO J. 2020, 39, e102209.
- Diaz, R.S.; Shytaj, I.L.; Giron, L.B.; Obermaier, B.; Della Libera, E.; Galinskas, J.; Dias, D.; Hunter, J.; Janini, M.; Gosuen, G.; et al. Potential impact of the antirheumatic agent auranofin on proviral HIV-1 DNA in individuals under intensified antiretroviral therapy: Results from a randomised clinical trial. Int. J. Antimicrob. Agents 2019, 54, 592–600.
- Valle-Casuso, J.C.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M.; et al. Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4+ T Cells and Offers an Opportunity to Tackle Infection. Cell Metab. 2019, 29, 611–626.e5.
- Castellano, P.; Prevedel, L.; Valdebenito, S.; Eugenin, E.A. HIV infection and latency induce a unique metabolic signature in human macrophages. Sci. Rep. 2019, 9, 1–14.
- Thaker, S.K.; Ch’Ng, J.; Christofk, H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019, 17, 1–15.
- Eisenreich, W.; Rudel, T.; Heesemann, J.; Goebel, W. How Viral and Intracellular Bacterial Pathogens Reprogram the Metabolism of Host Cells to Allow Their Intracellular Replication. Front. Cell. Infect. Microbiol. 2019, 9, 42.
- Timilsina, U.; Gaur, R. Modulation of apoptosis and viral latency—an axis to be well understood for successful cure of human immunodeficiency virus. J. Gen. Virol. 2016, 97, 813–824.
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26.
- Bobardt, M.; Kuo, J.; Chatterji, U.; Chanda, S.; Little, S.J.; Wiedemann, N.; Vuagniaux, G.; Gallay, P.A. The inhibitor apoptosis protein antagonist Debio 1143 Is an attractive HIV-1 latency reversal candidate. PLoS ONE 2019, 14, e0211746.
- Bobardt, M.; Kuo, J.; Chatterji, U.; Wiedemann, N.; Vuagniaux, G.; Gallay, P. The inhibitor of apoptosis proteins antagonist Debio 1143 promotes the PD-1 blockade-mediated HIV load reduction in blood and tissues of humanized mice. PLoS ONE 2020, 15, e0227715.
- Kuo, H.-H.; Ahmad, R.; Lee, G.Q.; Gao, C.; Chen, H.-R.; Ouyang, Z.; Szucs, M.J.; Kim, D.; Tsibris, A.; Chun, T.-W.; et al. Anti-apoptotic Protein BIRC5 Maintains Survival of HIV-1-Infected CD4+ T Cells. Immunity 2018, 48, 1183–1194.e5.
- Chugh, P.; Bradel-Tretheway, B.; Monteiro-Filho, C.M.; Planelles, V.; Maggirwar, S.B.; Dewhurst, S.; Kim, B. Akt inhibitors as an HIV-1 infected macrophage-specific anti-viral therapy. Retrovirology 2008, 5, 11.
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; D’Aloja, P.; Schürmann, A.; Baur, A.S. HIV-1 Nef associated PAK and PI3-Kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224.
- She, Q.-B.; Halilovic, E.; Ye, Q.; Zhen, W.; Shirasawa, S.; Sasazuki, T.; Solit, D.B.; Rosen, N. 4E-BP1 Is a Key Effector of the Oncogenic Activation of the AKT and ERK Signaling Pathways that Integrates Their Function in Tumors. Cancer Cell 2010, 18, 39–51.
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. mTOR Kinase Inhibition Causes Feedback-Dependent Biphasic Regulation of AKT Signaling. Cancer Discov. 2011, 1, 248–259.
- Campbell, G.R.; To, R.K.; Spector, S.A. TREM-1 Protects HIV-1-Infected Macrophages from Apoptosis through Maintenance of Mitochondrial Function. mBio 2019, 10.
- Chandrasekar, A.P.; Cummins, N.W.; Badley, A.D. The Role of the BCL-2 Family of Proteins in HIV-1 Pathogenesis and Persistence. Clin. Microbiol. Rev. 2019, 33.
- Doitsh, G.; Galloway, N.L.K.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Muñoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nat. Cell Biol. 2014, 505, 509–514.
- Zhang, G.; Luk, B.T.; Wei, X.; Campbell, G.R.; Fang, R.H.; Zhang, L.; Spector, S.A. Selective cell death of latently HIV-infected CD4+ T cells mediated by autosis inducing nanopeptides. Cell Death Dis. 2019, 10, 1–14.
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209.
- Wagner, R.N.; Reed, J.C.; Chanda, S.K. HIV-1 protease cleaves the serine-threonine kinases RIPK1 and RIPK. Retrovirology 2015, 12, 1–16.
- Fletcher-Etherington, A.; Nobre, L.; Nightingale, K.; Antrobus, R.; Nichols, J.; Davison, A.J.; Stanton, R.J.; Weekes, M.P. Human cytomegalovirus protein pUL36: A dual cell death pathway inhibitor. Proc. Natl. Acad. Sci. USA 2020, 117, 18771–18779.
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; Hashemi, M.; Glover, K.K.M.; Sher, A.A.; Coombs, K.M.; et al. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413.
- Swadling, L.; Pallett, L.J.; Diniz, M.O.; Baker, J.M.; Amin, O.E.; Stegmann, K.A.; Burton, A.R.; Schmidt, N.M.; Jeffery-Smith, A.; Zakeri, N.; et al. Human Liver Memory CD8+ T Cells Use Autophagy for Tissue Residence. Cell Rep. 2020, 30, 687–698.e6.
- He, X.; Yang, W.; Zeng, Z.; Wei, Y.; Gao, J.; Zhang, B.; Li, L.; Liu, L.; Wan, Y.; Zeng, Q.; et al. NLRP3-dependent pyroptosis is required for HIV-1 gp120-induced neuropathology. Cell. Mol. Immunol. 2020, 17, 283–299.
- Carvalho, A.R.; Pinto, C.M.; Tavares, J.N. Maintenance of the latent reservoir by pyroptosis and superinfection in a fractional order HIV transmission model. Int. J. Optim. Control. Theor. Appl. 2019, 9, 69–75.
- López-Huertas, M.R.; Morín, M.; Madrid-Elena, N.; Gutiérrez, C.; Jiménez-Tormo, L.; Santoyo, J.; Sanz-Rodríguez, F.; Pelayo, M.; Ángel, M.; Bermejo, L.G.; et al. Selective miRNA Modulation Fails to Activate HIV Replication in In Vitro Latency Models. Mol. Ther. Nucleic Acids 2019, 17, 323–336.
- Narayanan, A.; Iordanskiy, S.; Das, R.; Van Duyne, R.; Santos, S.; Jaworski, E.; Guendel, I.; Sampey, G.; Dalby, E.; Iglesias-Ussel, M.; et al. Exosomes Derived from HIV-1-infected Cells Contain Trans-activation Response Element RNA. J. Biol. Chem. 2013, 288, 20014–20033.
- Sampey, G.C.; Saifuddin, M.; Schwab, A.; Barclay, R.; Punya, S.; Chung, M.-C.; Hakami, R.M.; Zadeh, M.A.; Lepene, B.; Klase, Z.A.; et al. Exosomes from HIV-1-infected Cells Stimulate Production of Pro-inflammatory Cytokines through Trans-activating Response (TAR) RNA. J. Biol. Chem. 2016, 291, 1251–1266.
- Henderson, L.J.; Johnson, T.P.; Smith, B.R.; Reoma, L.B.; Santamaria, U.A.; Bachani, M.; DeMarino, C.; Barclay, R.A.; Snow, J.; Sacktor, N.; et al. Presence of Tat and transactivation response element in spinal fluid despite antiretroviral therapy. AIDS 2019, 33, S145–S157.
- Chen, L.; Feng, Z.; Yuan, G.; Emerson, C.C.; Stewart, P.L.; Ye, F.; Jin, G. Human Immunodeficiency Virus-Associated Exosomes Promote Kaposi’s Sarcoma-Associated Herpesvirus Infection via the Epidermal Growth Factor Receptor. J. Virol. 2020, 94.
- Barclay, R.A.; Schwab, A.; DeMarino, C.; Akpamagbo, Y.; Lepene, B.; Kassaye, S.; Iordanskiy, S.; Kashanchi, F. Exosomes from uninfected cells activate transcription of latent HIV. J. Biol. Chem. 2017, 292, 11682–11701.
- Barclay, R.A.; Mensah, G.A.; Cowen, M.; DeMarino, C.; Kim, Y.; Pinto, D.O.; Erickson, J.; Kashanchi, F. Extracellular Vesicle Activation of Latent HIV-1 Is Driven by EV-Associated c-Src and Cellular SRC-1 via the PI3K/AKT/mTOR Pathway. Viruses 2020, 12, 665.
- Chen, L.; Feng, Z.; Yue, H.; Bazdar, D.; Mbonye, U.; Zender, C.; Harding, C.V.; Bruggeman, L.; Karn, J.; Sieg, S.F.; et al. Exosomes derived from HIV-1-infected cells promote growth and progression of cancer via HIV TAR RNA. Nat. Commun. 2018, 9, 1–12.
- DeMarino, C.; Pleet, M.L.; Cowen, M.; Barclay, R.A.; Akpamagbo, Y.; Erickson, J.; Ndembi, N.; Charurat, M.; Jumare, J.; Bwala, S.; et al. Antiretroviral Drugs Alter the Content of Extracellular Vesicles from HIV-1-Infected Cells. Sci. Rep. 2018, 8, 1–20.
- DeMarino, C.; Cowen, M.; Pleet, M.L.; Pinto, D.O.; Khatkar, P.; Erickson, J.; Docken, S.S.; Russell, N.; Reichmuth, B.; Phan, T.; et al. Differences in Transcriptional Dynamics Between T-cells and Macrophages as Determined by a Three-State Mathematical Model. Sci. Rep. 2020, 10, 1–22.
- Sampey, G.C.; Iordanskiy, S.; Pleet, M.L.; DeMarino, C.; Romerio, F.; Mahieux, R.; Kashanchi, F. Identification of Modulators of HIV-1 Proviral Transcription from a Library of FDA-Approved Pharmaceuticals. Viruses 2020, 12, 1067.
- Dobrowolski, C.; Valadkhan, S.; Graham, A.C.; Shukla, M.; Ciuffi, A.; Telenti, A.; Karn, J.; Ott, M.; Henderson, A.; Spina, C. Entry of Polarized Effector Cells into Quiescence Forces HIV Latency. mBio 2019, 10, e00337-19.
- Theron, A.J.; Anderson, R.; Rossouw, T.M.; Steel, H.C. The Role of Transforming Growth Factor Beta-1 in the Progression of HIV/AIDS and Development of Non-AIDS-Defining Fibrotic Disorders. Front. Immunol. 2017, 8, 1461.
- Mbonye, U.; Wang, B.; Gokulrangan, G.; Shi, W.; Yang, S.; Karn, J. Cyclin-dependent kinase 7 (CDK7)-mediated phosphorylation of the CDK9 activation loop promotes P-TEFb assembly with Tat and proviral HIV reactivation. J. Biol. Chem. 2018, 293, 10009–10025.
- Giffin, M.J.; Stroud, J.C.; Bates, D.L.; Von Koenig, K.D.; Hardin, J.W.; Chen, L. Structure of NFAT1 bound as a dimer to the HIV-1 LTR κB element. Nat. Struct. Mol. Biol. 2003, 10, 800–806.
- Chan, J.K.; Bhattacharyya, D.; Lassen, K.G.; Ruelas, D.; Greene, W.C. Calcium/Calcineurin Synergizes with Prostratin to Promote NF-κB Dependent Activation of Latent HIV. PLoS ONE 2013, 8, e77749.
- Gerritsen, M.E.; Williams, A.J.; Neish, A.S.; Moore, S.; Shi, Y.; Collins, T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc. Natl. Acad. Sci. USA 1997, 94, 2927–2932.
- Kinoshita, S.; Su, L.; Amano, M.; Timmerman, A.L.; Kaneshima, H.; Nolan, G.P. The T Cell Activation Factor NF-ATc Positively Regulates HIV-1 Replication and Gene Expression in T Cells. Immunity 1997, 6, 235–244.
- Yang, X.; Chen, Y.; Gabuzda, D. ERK MAP Kinase Links Cytokine Signals to Activation of Latent HIV-1 Infection by Stimulating a Cooperative Interaction of AP-1 and NF-κB. J. Biol. Chem. 1999, 274, 27981–27988.
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nat. Cell Biol. 1987, 326, 711–713.
- Mbonye, U.; Karn, J. The Molecular Basis for Human Immunodeficiency Virus Latency. Annu. Rev. Virol. 2017, 4, 261–285.
- Cullen, B.R.; Lomedico, P.T.; Ju, G. Transcriptional interference in avian retroviruses—Implications for the promoter insertion model of leukaemogenesis. Nat. Cell Biol. 1984, 307, 241–245.
- Lenasi, T.; Contreras, X.; Peterlin, B.M. Transcriptional Interference Antagonizes Proviral Gene Expression to Promote HIV Latency. Cell Host Microbe 2008, 4, 123–133.
- Colin, L.; Van Lint, C. Molecular control of HIV-1 postintegration latency: Implications for the development of new therapeutic strategies. Retrovirology 2009, 6, 1–29.
- Greger, I.H. Transcriptional interference perturbs the binding of Sp1 to the HIV-1 promoter. Nucleic Acids Res. 1998, 26, 1294–1301.
- Renner, D.B.; Yamaguchi, Y.; Wada, T.; Handa, H.; Price, D.H. A Highly Purified RNA Polymerase II Elongation Control System. J. Biol. Chem. 2001, 276, 42601–42609.
- Pagano, J.M.; Kwak, H.; Waters, C.T.; Sprouse, R.O.; White, B.S.; Ozer, A.; Szeto, K.; Shalloway, D.; Craighead, H.G.; Lis, J.T. Defining NELF-E RNA Binding in HIV-1 and Promoter-Proximal Pause Regions. PLoS Genet. 2014, 10, e1004090.
- Kulkarni, S.; Lied, A.; Kulkarni, V.; Rucevic, M.; Martin, M.P.; Walker-Sperling, V.; Anderson, S.K.; Ewy, R.; Singh, S.; Nguyen, H.; et al. CCR5AS lncRNA variation differentially regulates CCR5, influencing HIV disease outcome. Nat. Immunol. 2019, 20, 824–834.
- Rao, S.; Amorim, R.; Niu, M.; Breton, Y.; Tremblay, M.J.; Mouland, A.J. Host mRNA decay proteins influence HIV-1 replication and viral gene expression in primary monocyte-derived macrophages. Retrovirology 2019, 16, 1–15.

