 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Paolo Bollella | + 2554 word(s) | 2554 | 2020-04-21 07:46:55 | | | |
| 2 | Catherine Yang | Meta information modification | 2554 | 2020-05-13 11:00:20 | | | | |
| 3 | Catherine Yang | -1 word(s) | 2553 | 2020-05-13 11:02:07 | | | | |
| 4 | Catherine Yang | -52 word(s) | 2501 | 2020-10-28 10:06:37 | | |
Video Upload Options
Porous gold (PG) layers modified electrodes have emerged as valuable enzyme support to realize multiple enzyme-based bioelectrochemical devices like biosensors, enzymatic fuel cells (EFCs), smart drug delivery devices triggered by enzyme catalyzed reactions, etc. PG films can be synthesized by using different methods such as dealloying, electrochemical (e.g., templated electrochemical deposition, self-templated electrochemical deposition etc.) self-assembly and sputter deposition. Herein, all the recent findings about PG enzyme based-electrodes for biosensors and enzymatic fuel cells (EFCs) development are discussed.
All the recent findings about PG enzyme based-electrodes for biosensors and enzymatic fuel cells (EFCs) development are discussed have been summarized.
1. Dehydrogenases
Recently, one- and multi-cofactor dehydrogenases like flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN) and pyrroloquinoline quinone (PQQ) dependent dehydrogenases, containing sometimes an additional heme-based electron transfer subunit, were demonstrated that can undergo direct electron transfer (DET) at several different electrode platforms [1][2][3]. In particular, Siepenkoetter and his coworkers reported on the covalent immobilization of fructose dehydrogenase (FDH) onto nano-PG electrode, as shown in Figure 1.
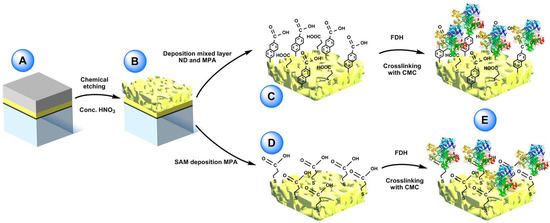
Figure 1. (A) Sputtered glass sheet with layers of titanium, pure gold and gold/silver alloy (bottom to top). (B) nano-PG electrode surface post-dealloying by concentrated nitric acid. (C) Electrochemical reduction of 6-amino-2-Naphthoic diazonium salt (ND) on the nano-PG surface utilizing a single scan and subsequent filling of the void spaces with 3-mercaptopropionic acid (MPA). (D) Preparation of mercaptopropionic acid (MPA) SAM on nano-PG substrate by immersion over night at 4 °C. (E) After adsorption of FDH on the two modified electrodes the enzyme was crosslinked using N-cyclohexyl-N’-(2-morpholinoethyl) carbodiimide metho-p-toluenesulfonate (CMC). Reproduced with permission from [2]. Copyright Wiley, 2017.
FDH is a heterotrimeric membrane-bound enzyme complex, consisting of three subunits, especially subunit I (DHFDH), which is the catalytic domain where D-fructose is involved in a 2H+/2e− oxidation to produce 5-dehydro-d-fructose, and subunit II (CYTFDH), which acts as a built-in electron transfer unit between DHFDH and the electrode surface.[3][4][5] In this paper, nano-PG was reported to be prepared through chemical dealloying of silver-gold alloy by using different temperatures and dealloying times in order to obtain different pore size. Nano-PG electrodes with average pore sizes ranging from 9 to 62 nm have been studied.[6] Indeed, the nano-PG electrode with an average pore size of 42 nm produced the highest current densities. This platform was further used to develop a DET biosensor for the detection of fructose exhibiting a sensitivity of 3.7 ± 0.2 µA cm−2 mM−1, a limit of detection of 1.2 µM and an increased high thermal (up to 35 °C) and storage stability (40% of initial activity retained after 6 days) mainly due to the porous structure protecting the immobilized enzyme.
In our previous paper, we reported a self-templated electrodeposition of highly-PG obtained with a two-step method.[7] The so nanostructured electrode was further modified with different self-assembled monolayers owing different functional groups, namely –COOH (4-mercaptobenzoic acid), –OH (4-mercaptophenol) and –NH2 (4-aminothiophenol) in order to produce differently charged electrode surface for an effective immobilization of FDH through electrostatically driven adsorption. The electrode modified with 4-mercaptophenol exhibited the largest catalytic current density (about 1000 μA cm−2 in the presence of 10 mM fructose) due to the presence of –OH groups on the electrode surface, which stabilize and orientate the enzyme layer on the electrode surface. The so modified electrode displayed excellent analytical features such as a linear range between 0.05 and 5 mM, a sensitivity of 175 ± 15 μA cm−2 mM−1, a detection limit of 0.3 µM, an increased high thermal (up to 35 °C) and an extraordinary storage stability (90% retained activity over 90 days). In this case, the protection of the enzyme layer is ensured from the large pores (almost 10 µm in diameter) and from their nanostructures where the high number of enzyme molecules is effectively retained.
However, we should consider several examples where dehydrogenases have been immobilized onto PG base electrodes in a mediated electron transfer (MET) architecture. In this regard, Ben-Ali et al. reported the development of a novel electrochemical method for the deposition of highly ordered microporous gold nanostructure by using colloidal lattice as templates because they can be produced with an almost perfect arrangement.[8] Afterwards, the colloidal lattice is removed by using an appropriate solvent, which resulted in a really ordered microporous structure able to support the mediated electron transfer of nicotinamide adenine dinucleotide (NAD)-dependent glucose dehydrogenase (GDH).[9] The electrode surface was further modified by using the layer-by-layer method alternating the mediator, the NAD+ cofactor and the enzyme stabilized from the Nafion® membrane. It should also be mentioned the presence of an intermediate layer of Ca2+ between the mediator and NAD+, which is well known to enhance the kinetics of the electron transfer between NAD+ and the mediator. This supramolecular electrode architecture showed great electrocatalytic activity towards glucose oxidation at relatively low overpotential (almost −100 mV vs. SCE).
In a similar approach, Szamocki and his coworkers reported the immobilization of NAD-dependent GDH onto highly ordered macroporous gold electrode modified with (4-carboxy-2,5,7-trinitro-9-fluorenylidene)malononitrile (TNF) and NAD+. The enzyme was cross-linked by using glutaraldehyde. GDH/NAD+/TNF/PG electrode displayed great electrocatalytic cyclic voltammograms in presence of glucose.[10]
Moreover, Xiao et al. reported the development of a symmetric supercapacitor/biofuel cell based on the electrodeposition of a silver/gold alloy further chemically dealloyed. This PG electrode preparation method has been already deeply discussed in the previous sections. In this study, they were able to support the MET of flavin adenine dinucleotide (FAD)-dependent GDH through the electrodeposited poly(3,4-ethylenedioxythiophene) (PEDOT) and the redox polymer [Os(2,2′-bipyridine)2(polyvinylimidazole)10Cl]+/2+(Os(bpy)2PVI). The same electrode construction method has been employed for the immobilization of the cathodic enzyme, namely bilirubin oxidase (BOx). This hybrid device allowed us to store in the supercapacitor the energy yielded by the biofuel cell with an effective delivery of a high power pulse. Notably, it released a pulse current density of 2 mA cm−2, which correspond to an instant maximum power density of 609 μW cm−2 (468 times higher than that of the EFC).[11]
Moreover, Salaj-Kosla et al. reported the immobilization of cellobiose dehydrogenase (CDH) [12] onto a similarly prepared PG electrode. The enzyme was covalently cross-linked to an osmium-based redox polymer supporting its MET. [13][14] The so modified electrode showed great catalytic current towards lactose oxidation and an exceptionally poor operating stability loosing 70% of the osmium redox polymer initially immobilized onto the electrode.
In our previous paper, we reported the self-templated electrodeposition of PG gold onto microneedle-based electrodes [15] developing a minimally invasive biosensor for the detection of glucose based on MET of FAD-dependent GDH.[16][17] The highly porous gold surface of the microneedles was further modified by using 6-(ferrocenyl)hexanethiol (FcSH) as a redox mediator and drop-casting the FAD-GDH enzyme solution, as schematically reported in Figure 2A,B. The microneedle-based FcSH/FAD-GDH biosensor showed an extended linear range (0.1–10 mM), high sensitivity (50.86 µA cm−2 mM−1) and good storage stability (20% signal loss after 30 days).
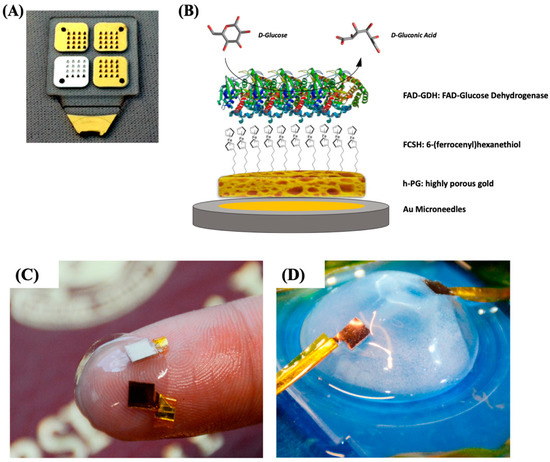
Figure 2. (A) Photo of four 4 × 4 microneedle arrays metallized with gold and silver and (B) schematic representation of the highly porous gold (h-PG)/6-(ferrocenyl)hexanethiol (FcSH)/flavin adenine dinucleotide glucose dehydrogenase (FAD-GDH)/Au microneedle-based glucose biosensor. Photograph of the contact lens encapsulated enzymatic fuel cell (EFC; C) and testing setup (D). Figure 2A,B are reproduced with permission from [15]. Copyright MDPI, 2019. Figure 2C,D are reproduced with permission from [18]. Copyright American Chemical Society, 2018.
2. Oxidases
The group of oxidases is another family of redox enzymes that has been successfully immobilized onto PG based electrodes. Oxidases have been widely employed for the development of first generation (oxidation current is mainly related with the oxidation/reduction of H2O2 at the electrode) and second generation biosensors (the electrons are shuttled from a mediator to the electrode).Du Toit and coworkers [21] reported on the potentiostatic self-templated electrodeposition of highly PG layer by using two steps: (i) small hydrogen bubbling by applying −0.7 V vs. SCE and (ii) a vigorous hydrogen bubbling by applying −4 V vs. SCE. Afterwards, the authors explored two different immobilization methods, one based on the electrodeposition of glucose oxidase (GOx) by cycling the electrode between 0.42 and 0.6 V vs. SCE, and the other one by simple adsorption of the enzyme onto the electrode surface. The first method resulted in a more intimate contact between the redox enzyme and the pore internal surface enhancing by approximately 6 times the glucose oxidation current compared to the electrode obtained through enzyme adsorption. The so modified electrode has been reported either as first generation biosensor for glucose detection and as a bioanode of EFC.
Moreover, Szamocky et al. [22] reported on GOx immobilization onto a highly ordered macroporous electrode by incorporating the enzyme into an electrodeposition paint (EDP). The electrodeposition paint is a copolymer of acrylic acid and various acrylates (water soluble in the original form). By applying a sufficiently positive voltage to oxidize water, the pH decreases locally, therefore the acrylic acid gets protonated forming a thin layer on the electrode and incorporating GOx. The so prepared electrode showed great catalytic properties towards glucose oxidation, which is detected through H2O2 electrochemical oxidation at the electrode surface.
Furthermore, Sanzò et al. reported the immobilization of GOx onto a one-step self-templated PG electrode for the development of a first generation glucose biosensor. GOx was easily immobilized through covalent cross-linking with glutaraldehyde. The so modified electrode showed a higher sensitivity of 48.3 ± 0.9 μA cm−2 mM−1 at 0.45 V vs. SCE compared to a value of 24.6 ± 1.3 μA cm−2 mM−1 at 0.70 V vs. SCE obtained with bare Au electrodes.[23]
Xiao et al. [20] reported the immobilization of lactate oxidase (LOx) onto nano-PG electrode (obtained through electrodeposition and successive chemical dealloying) for the development of EFC onto contact lenses. After the preliminary step, the PG electrode was further modified with an osmium redox polymer in order to promote the MET of LOx, while BOx was immobilized in direct electron transfer.[24] Once prepared, the electrodes were placed between two contact lenses in order to obtain a flexible EFC, as displayed in Figure 2C,D. The EFC showed a maximum power density of 1.7 ± 0.1 μW cm−2 and an open-circuit voltage of 380 ± 28 mV in air-equilibrated artificial tears solution (containing approximately 3 mM lactate). A good EFC operational stability could be observed retaining 20% of its initial power output after 5.5 h maybe due to the protective effect of porous nanostructured electrodes.
Heme Proteins
Cytochrome c (cyt c) has been widely used as a model protein that can undergo direct electron transfer onto self-assembled monolayer (SAM) modified electrode.[19] Cyt c electrochemical behavior is greatly affected by the topography of gold electrodes (enzyme immobilized through easy drop-casting). In particular, it was demonstrated that highly rough gold surfaces could result in SAMs containing multiple defects, ensuring an increased cyt c surface coverage. A PG electrode modified with cyt c covalently immobilized onto a mixed SAM showed a well-defined and almost symmetric voltammogram (peak-to-peak separation ΔEp = 18 mV). The surface coverage of active cyt c onto the modified electrode was approximately 11 times higher than that achieved at a planar Au surface. The so prepared electrode showed an extremely high operating stability with no decreases in the voltammetric response under continuous potential cycling (30 scans) in an aqueous buffer.
3. Multicopper Oxidases (MCOs)
Multicopper oxidases group (MCOs, e.g., laccase and bilirubin oxidase (BOx)) includes many enzymes able to reduce dioxygen to water (four electrons) with the contemporary one-electron oxidation of an electron donor (e.g., catechol, 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS), etc.).[26] MCOs modified electrodes have been mainly used as biocathodes of EFCs.[27] MCOs contain four copper atoms: the T1 copper site acts as an electron acceptor (DET configuration: electrons donated from the electrode and MET configuration: electrons donated from a diffusive or immobilized mediator) while O2 is reduced to H2O at the T2/T3 trinuclear copper cluster.[28][29] In this regard, Lopez and coworkers reported the immobilization of BOx onto nano-PG electrodes by using a potential pulse method.[30] The potentials applied were selected based on the potential of zero charge (pzc), which resulted to be different for a polycrystalline gold electrode and PG electrodes with different morphology, as reported in Figure 3. Indeed, the latter resulted to be the critical factor for the determination of potential needed for the enzyme deposition. This method allowed a selective orientation of BOx with the T1 site toward the electrode in order to be able to accept electrons directly from the electrode (DET).[31] Thus, the high values of current density related to dioxygen reduction could mainly be attributed to the enhanced enzyme surface coverage and to the efficient enzyme orientation.
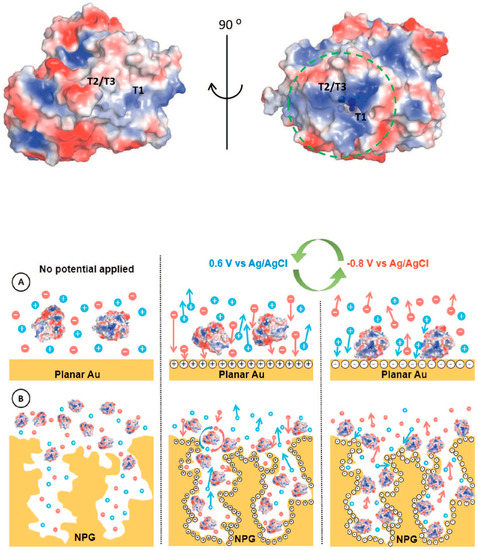
Figure 3. Surface charge distribution pattern of (PBD 2XLL) Myrothecium verrucaria BOx (blue and red colors symbolize positive and negative charges, respectively). Potential pulse-assisted immobilization approach at (A) planar gold and (B) nano-PG electrodes. The green arrows indicate the changes in the potential pulses of below and above pzc values. Reproduced with permission from [20]. Copyright Elsevier Ltd., 2018.
In a similar approach, Salaj-Kosla et al. reported the immobilization of high potential laccase, namely Thrametes hirsuta laccase, onto nano-PG electrode.[21] In this case, the immobilization was initially performed through adsorption of the enzyme into the porous structure, resulting in poor catalytic performance and a continuous leakage of the enzyme from the electrode. Thus, the electrode surface was later stabilized by using epoxy-based polymers able to cross-link the enzyme into the porous structure. However, the authors investigated also the resistivity of the enzyme modified electrode towards two different inhibitors, namely fluoride and chloride. It should be mentioned that fluoride acts as a non-competitive inhibitor by binding to the T2/T3 copper cluster, blocking the ET pathway from the T1 site to the T2/T3 cluster. This results in the absence of the electrocatalytic wave typical for dioxygen reduction, even with a minimal presence of fluoride. Conversely, chloride is considered a competitive inhibitor, which hinders the electron pathway between the electron donors and the T1 site (DET: electrodes; MET: diffusive or immobilized mediator). In the latter case, the nano-PG electrode was able to retain 50% of its initial activity after the addition of 150 mM of Cl−, probably due to the intimate contact between the enzyme and the pore surface.[22] Moreover, the electrode showed also a good thermal stability gaining 40% of activity by increasing the temperature up to 37 °C. The last feature of this electrode opens some future avenue for its employment in implantable devices. Moreover, the electrodes reported above have been successfully used to develop glucose/O2 or lactate/O2 EFCs.[23]
4. Conclusions
PG electrodes have been proven to be a promising support for enzyme immobilization improving their response and stability towards multiple enzyme-based bioelectrochemical applications (e.g., biosensors and biofuel cells). Notably, PG modified electrodes exhibit four main advantages towards the development of enzyme-based devices, namely increasing stability of the immobilized enzyme, enhanced electron transfer reaction rate, increasing stability of self-assembled monolayers (SAMs) and hindering of surface biofouling. In particular, it was demonstrated that PG layers can increase the stability of enzyme layers when exposed to high temperatures and organic solvents. Moreover, PG electrodes enhanced the stability of SAMs allowing a stable covalent linking of redox enzymes. Finally, it was clearly demonstrated the possibility to drastically increase the ET rate constants of immobilized redox enzymes as well as good anti-biofouling properties.
References
- Paolo Bollella; Porous Gold: A New Frontier for Enzyme-Based Electrodes. Nanomaterials 2020, 10, 722, 10.3390/nano10040722.
- Paolo Bollella; Lo Gorton; Riccarda Antiochia; Direct Electron Transfer of Dehydrogenases for Development of 3rd Generation Biosensors and Enzymatic Fuel Cells. Sensors 2018, 18, 1319, 10.3390/s18051319.
- Paolo Bollella; Yuya Hibino; Paolo Conejo-Valverde; Jackeline Soto-Cruz; Julian Bergueiro; Marcelo Calderón; Oscar Rojas-Carrillo; Kenji Kano; Lo Gorton; The influence of the shape of Au nanoparticles on the catalytic current of fructose dehydrogenase. Zeitschrift für Analytische Chemie 2019, 411, 7645-7657, 10.1007/s00216-019-01944-6.
- Paolo Bollella; Yuya Hibino; Kenji Kano; Lo Gorton; Riccarda Antiochia; Enhanced Direct Electron Transfer of Fructose Dehydrogenase Rationally Immobilized on a 2-Aminoanthracene Diazonium Cation Grafted Single-Walled Carbon Nanotube Based Electrode. ACS Catalysis 2018, 8, 10279-10289, 10.1021/acscatal.8b02729.
- Paolo Bollella; Yuya Hibino; Kenji Kano; Lo Gorton; Riccarda Antiochia; The influence of pH and divalent/monovalent cations on the internal electron transfer (IET), enzymatic activity, and structure of fructose dehydrogenase. Zeitschrift für Analytische Chemie 2018, 410, 3253-3264, 10.1007/s00216-018-0991-0.
- Till Siepenkoetter; Urszula Salaj-Kosla; Xinxin Xiao; Serguei Belochapkine; Edmond Magner; Nanoporous Gold Electrodes with Tuneable Pore Sizes for Bioelectrochemical Applications. Electroanalysis 2016, 28, 2415-2423, 10.1002/elan.201600249.
- Paolo Bollella; Yuya Hibino; Kenji Kano; Lo Gorton; Riccarda Antiochia; Highly Sensitive Membraneless Fructose Biosensor Based on Fructose Dehydrogenase Immobilized onto Aryl Thiol Modified Highly Porous Gold Electrode: Characterization and Application in Food Samples. Analytical Chemistry 2018, 90, 12131-12136, 10.1021/acs.analchem.8b03093.
- Samia Ben Ali; David A. Cook; Philip N. Bartlett; Alexander Kühn; Bioelectrocatalysis with modified highly ordered macroporous electrodes. Journal of Electroanalytical Chemistry 2005, 579, 181-187, 10.1016/j.jelechem.2004.11.018.
- Paul Kavanagh; Dónal Leech; Mediated electron transfer in glucose oxidising enzyme electrodes for application to biofuel cells: recent progress and perspectives. Physical Chemistry Chemical Physics 2012, 15, 4859, 10.1039/c3cp44617d.
- Rafael Szamocki; Alexandra Velichko; Frank Mücklich; Stéphane Reculusa; Serge Ravaine; Sebastian Neugebauer; Wolfgang Schuhmann; Rolf Hempelmann; Alexander Kuhn; Improved enzyme immobilization for enhanced bioelectrocatalytic activity of porous electrodes. Electrochemistry Communications 2007, 9, 2121-2127, 10.1016/j.elecom.2007.06.008.
- Xinxin Xiao; Peter Ó Conghaile; Dónal Leech; Roland Ludwig; Edmond Magner; A symmetric supercapacitor/biofuel cell hybrid device based on enzyme-modified nanoporous gold: An autonomous pulse generator. Biosensors and Bioelectronics 2017, 90, 96-102, 10.1016/j.bios.2016.11.012.
- Urszula Salaj-Kosla; Micheál D. Scanlon; Tobias Baumeister; Kawah Zahma; Roland Ludwig; Peter Ó Conghaile; Domhnall Mac Aodha; Dónal Leech; Edmond Magner; Mediated electron transfer of cellobiose dehydrogenase and glucose oxidase at osmium polymer-modified nanoporous gold electrodes. Zeitschrift für Analytische Chemie 2012, 405, 3823-3830, 10.1007/s00216-012-6657-4.
- Adam Heller; Electron-conducting redox hydrogels: design, characteristics and synthesis. Current Opinion in Chemical Biology 2006, 10, 664-672, 10.1016/j.cbpa.2006.09.018.
- Adam Heller; Ben Feldman; Electrochemical Glucose Sensors and Their Applications in Diabetes Management. Chemical Reviews 2008, 108, 2482-2505, 10.1021/cr068069y.
- Paolo Bollella; Sanjiv Sharma; Anthony E. G. Cass; Federico Tasca; Riccarda Antiochia; Minimally Invasive Glucose Monitoring Using a Highly Porous Gold Microneedles-Based Biosensor: Characterization and Application in Artificial Interstitial Fluid. Catalysts 2019, 9, 580, 10.3390/catal9070580.
- Paolo Bollella; Sanjiv Sharma; Anthony Edward George Cass; Riccarda Antiochia; Minimally-invasive Microneedle-based Biosensor Array for Simultaneous Lactate and Glucose Monitoring in Artificial Interstitial Fluid. Electroanalysis 2019, 31, 374-382, 10.1002/elan.201800630.
- Joseph Wang; Electrochemical Glucose Biosensors. Chemical Reviews 2008, 108, 814-825, 10.1021/cr068123a.
- Gabriella Sanzò; Irene Taurino; Riccarda Antiochia; Lo Gorton; Gabriele Favero; Franco Mazzei; Giovanni De Micheli; Sandro Carrara; Bubble electrodeposition of gold porous nanocorals for the enzymatic and non-enzymatic detection of glucose. Bioelectrochemistry 2016, 112, 125-131, 10.1016/j.bioelechem.2016.02.012.
- Michael C. Leopold; Edmond F. Bowden; Influence of Gold Substrate Topography on the Voltammetry of CytochromecAdsorbed on Carboxylic Acid Terminated Self-Assembled Monolayers. Langmuir 2002, 18, 2239-2245, 10.1021/la011456c.
- Francesca Lopez; Till Siepenkoetter; Xinxin Xiao; Edmond Magner; Wolfgang Schuhmann; Urszula Salaj-Kosla; Potential pulse-assisted immobilization of Myrothecium verrucaria bilirubin oxidase at planar and nanoporous gold electrodes. Journal of Electroanalytical Chemistry 2018, 812, 194-198, 10.1016/j.jelechem.2017.12.023.
- Urszula Salaj-Kosla; Sascha Poeller; Yvonne Beyl; Micheál D. Scanlon; Sergey Beloshapkin; Sergey Shleev; Wolfgang Schuhmann; Edmond Magner; Direct electron transfer of bilirubin oxidase (Myrothecium verrucaria) at an unmodified nanoporous gold biocathode. Electrochemistry Communications 2012, 16, 92-95, 10.1016/j.elecom.2011.12.007.
- Anne De Poulpiquet; Christian H. Kjaergaard; Jad Rouhana; Ievgen Mazurenko; Pascale Infossi; Sébastien Gounel; Roger Gadiou; Marie Thérèse Giudici-Orticoni; Edward I. Solomon; Nicolas Mano; et al.Elisabeth Lojou Mechanism of Chloride Inhibition of Bilirubin Oxidases and Its Dependence on Potential and pH. ACS Catalysis 2017, 7, 3916-3923, 10.1021/acscatal.7b01286.
- Magnus Falk; Zoltan Blum; Sergey Shleev; Direct electron transfer based enzymatic fuel cells. Electrochimica Acta 2012, 82, 191-202, 10.1016/j.electacta.2011.12.133.
- Magnus Falk; Viktor Andoralov; Zoltan Blum; Javier Sotres; Dmitry B. Suyatin; Tautgirdas Ruzgas; Thomas Arnebrant; Sergey Shleev; Biofuel cell as a power source for electronic contact lenses. Biosensors and Bioelectronics 2012, 37, 38-45, 10.1016/j.bios.2012.04.030.
- Michael C. Leopold; Edmond F. Bowden; Influence of Gold Substrate Topography on the Voltammetry of CytochromecAdsorbed on Carboxylic Acid Terminated Self-Assembled Monolayers. Langmuir 2002, 18, 2239-2245, 10.1021/la011456c.
- Sergey Shleev; Jan Tkac; Andreas Christenson; Tautgirdas Ruzgas; Alexander Yaropolov; James W. Whittaker; Lo Gorton; Direct electron transfer between copper-containing proteins and electrodes. Biosensors and Bioelectronics 2005, 20, 2517-2554, 10.1016/j.bios.2004.10.003.
- Paolo Bollella; Giovanni Fusco; Daniela Stevar; Lo Gorton; Roland Ludwig; Su Ma; Harry Boer; Anu Koivula; Cristina Tortolini; Gabriele Favero; et al.Riccarda AntiochiaFranco Mazzei A Glucose/Oxygen Enzymatic Fuel Cell based on Gold Nanoparticles modified Graphene Screen-Printed Electrode. Proof-of-Concept in Human Saliva. Sensors and Actuators B: Chemical 2018, 256, 921-930, 10.1016/j.snb.2017.10.025.
- Sergey Shleev; Asma El Kasmi; Tautgirdas Ruzgas; Lo Gorton; Direct heterogeneous electron transfer reactions of bilirubin oxidase at a spectrographic graphite electrode. Electrochemistry Communications 2004, 6, 934-939, 10.1016/j.elecom.2004.07.008.
- Dmitri Ivnitski; Constantine Khripin; Heather R. Luckarift; Glenn R. Johnson; Plamen Atanassov; Surface characterization and direct bioelectrocatalysis of multicopper oxidases. Electrochimica Acta 2010, 55, 7385-7393, 10.1016/j.electacta.2010.07.026.
- Francesca Lopez; Till Siepenkoetter; Xinxin Xiao; Edmond Magner; Wolfgang Schuhmann; Urszula Salaj-Kosla; Potential pulse-assisted immobilization of Myrothecium verrucaria bilirubin oxidase at planar and nanoporous gold electrodes. Journal of Electroanalytical Chemistry 2018, 812, 194-198, 10.1016/j.jelechem.2017.12.023.
- Huajun Qiu; Caixia Xu; Xirong Huang; Yi Ding; Yinbo Qu; Peiji Gao; Adsorption of Laccase on the Surface of Nanoporous Gold and the Direct Electron Transfer between Them. The Journal of Physical Chemistry C 2008, 112, 14781-14785, 10.1021/jp805600k.


