 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Riccardo Cristofani | + 2821 word(s) | 2821 | 2021-03-03 10:12:36 | | | |
| 2 | Peter Tang | Meta information modification | 2821 | 2021-03-10 03:43:10 | | | | |
| 3 | Peter Tang | Meta information modification | 2821 | 2021-03-10 03:43:42 | | |
Video Upload Options
The Heat Shock Protein B8 (HSPB8) is a small chaperone involved in chaperone-assisted selective autophagy (CASA). CASA promotes the selective degradation of proteins to counteract cell stress such as tumor-induced stress. HSPB8 is also involved in (i) the cell division machinery regulating chromosome segregation and cell cycle arrest in the G0/G1 phase and (ii) inflammation regulating dendritic cell maturation and cytokine production. HSPB8 expression and role are tumor-specific, showing a dual and opposite role.
1. Introduction
Human cells have different fine-tuned systems that act as defense mechanisms against a large number of different environmental stresses. Among these, one identified in the 1960s by Ferruccio Ritossa is the heat shock response (HSR), an extraordinary mechanism shared by procaryotes and eukaryotes which responds to and counteracts several potentially harmful cell stresses. In fact, the HSR is now identified more generally as a stress response, being triggered in different ways by a variety of stressful conditions affecting the cells. The stress response may arise outside (i.e., heat shock) or inside (i.e., protein misfolding) the cells, and it is based on the interplay between the nucleus and organelles such as mitochondria, the endoplasmic reticulum, autophagosomes, and lysosomes as well as membrane-less organelles such as P bodies and stress granules (SGs). The stress response is based on several cellular processes such as gene expression, protein synthesis, and protein and organelle degradation and it is involved in aging and in many human diseases such as neurodegeneration, inflammation, obesity, diabetes, autoimmune diseases, atherosclerosis, and cancer [1][2].
The stress response, including the HSR, is based on a rapid and transient gene expression mechanism that controls the expression of molecular chaperones: the heat shock proteins (HSPs). The six molecular chaperone families are ubiquitous and broadly conserved; even if not all of them are induced by heat shock, they are, indeed, differentially controlled by different stresses. Chaperones were originally grouped based on their apparent molecular mass, while now, they are classified mainly by their functions in the folding processes (HSP110s, HSP90s, HSP70s, HSP60s, HSP40s, and small heat shock proteins (sHSPs)), but there are similarities between the two nomenclatures; in fact, based on the HUGO Gene Nomenclature Committee, the new classification recently adopted is as follows: HSPB (sHSP), DNAJ (HSP40), HSPD (HSP60), HSPA (HSP70), HSPC (HSP90), and HSPH (HSP110) [3], and reflects both the functions and the sizes of the members of each subfamily.
The human HSPB8 is a member of the HSPBs which is a ubiquitous family of ATP-independent stress proteins, whose activity is mediated by other ATP-dependent chaperones (i.e., HSPAs), defining whether HSPBs clients can be refolded or degraded [4][5]. HSPBs are able to bind unfolded/misfolded substrates and subsequently new and larger oligomers are formed, preventing irreversible client aggregation and allowing ATP-dependent chaperones recognition to assist protein refolding [6]. HSPBs contribute to protein quality control (PQC) and work to prevent protein aggregation and to generate a pool of non-native proteins that can be rapidly folded [7]. The human genome encodes 10 HSPBs named HSPB1 through HSPB10, with an apparent molecular mass of 12–43 kDa [8]. Despite their name, the expression of HSPBs can be dependently or independently regulated by heat shock transcription factors (HSFs). In fact, it relies on the combinatory effects of many transcription factors. The basal expression or stress inducibility of HSPBs is therefore regulated by different cis-elements localized in the HSPB regulatory regions [9].
A common key feature of HSPBs is the alpha-crystallin domain (ACD), which refers to the best-known family member, the eye lens protein alpha-crystallin [10]. The ACD is flanked by a variable N-terminal domain (NTD) and a short C-terminal domain (CTD), which show high heterogeneity both in sequence and size among the HSPBs. All the three domains are involved in determining the quaternary structure adopted by the HSPBs. Indeed, while the ACD mediates the dimerization of HSPBs, the ACD flanking regions affect HSPBs oligomerization. Since the NTD and CTD highly differ among the HSPBs, the size and composition of HSPB oligomers can vary, ranging from mainly dimers to 600 kDa hetero-oligomers [11][12][13]. A dynamic association/dissociation has been suggested as a primary regulator of HSPB functions and it is often induced by protein phosphorylation [14].
HSPB8, also named small stress protein-like protein (sHSP22), protein kinase H11(H11), E2-induced gene 1 protein (E2IG1), or alpha-crystallin C chain (CRYAC), is different from other HSPBs, since it is preferentially found as monomers and homodimers, even if the protein can also interact with other HSPBs forming heterodimers [15][16][17]. In addition, like HSPB5 and HSPB6, HSPB8 represents an “atypical” member of the HSPB family because in mammalian cells, it can form stable complexes with the Bcl2-associated athanogene 3 (BAG3), which is thought to be the obligate partner of HSPB8 [18][19].
2. Tissue Distribution of HSPB8
In basal conditions, the heart and the skeletal muscle are the two most noteworthy tissues in which HSPB8 is expressed at relatively high levels. However, these tissues also express many other HSPBs, such as HSPB1, HSPB2, HSPB3, HSPB5, HSPB6, and HSPB7, possibly because during life, muscle tissues are subjected to several stressful conditions (including prolonged mechanical stress, hypoxia, damaged protein exposure). HSPB8 is also highly expressed in the brain, where it can be found both in neurons and in glial cells. Neuronal HSPB8 expression appears to be particularly relevant as a neuroprotective mechanism; for example, under stressful conditions, such as proteotoxic stresses, motoneurons express a very high level of HSPB8 in an attempt to protect themselves from damage [20]. Lower HSPB8 expression has been observed in the prostate, placenta, lungs, kidneys, and skin, while the ovaries, testes, liver, pancreas, and spleen appear to be devoid of HSPB8 [21]. As with many other HSPs, HSPB8 expression can be triggered by a variety of stresses, including starvation, proteasome inhibition, misfolded proteins accumulation, nuclear factor-Kappa B (NF-κB) activation, autophagy activation, microtubule destabilization, and occasionally heat shock [20][22][23][24][25][26].
3. Structure of HSPB8
HSPB8 (Figure 1A) is composed of 196 amino acids with an apparent molecular mass of 21.6 kDa. Its ACD is located close to the CTD approximately between amino acids 86 and 176. The short CTD interacts with aberrantly exposed hydrophobic regions of the substrate, preventing its aggregation during chaperone activity. Different from other members of the HSPB family (HSPB1, HSPB2, HSPB4, and HSPB5), HSPB8 does not contain the conserved I/V-X-I/V motif responsible for oligomeric assembly [27][28]. Indeed, this motif interacts with the neighboring ACD in a pocket between strands β4 and β8, acting as a bridge between the dimers. Instead, a hydrophobic pocket mediates the HSPB8 binding to two I/V-X-I/V domains, namely, IPV domains, present in the BAG3 protein [29], thus explaining the preferential binding of HSPB8 to BAG3 rather than HSPB8 assembly into large oligomers. In this interaction, two molecules of HSPB8 complement one BAG3 molecule, forming a functional chaperone complex (see below).
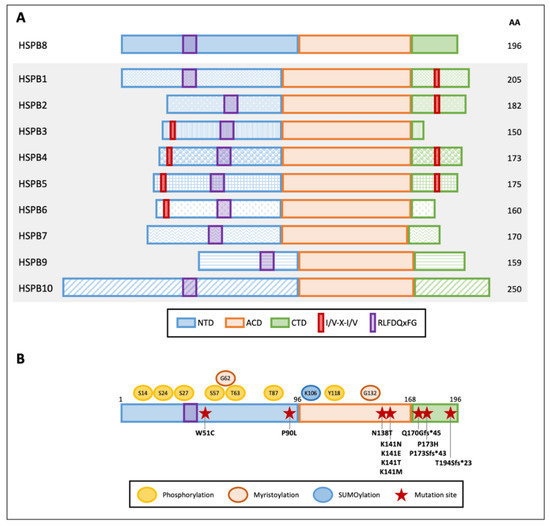
Figure 1. Graphical representation of human small heat shock proteins (HSPBs) structure and features. (A) Schematic diagram of the domains within human HSPBs. NTD and CTD indicate the variable N- and C-terminal domains, respectively; ACD indicates the conserved alpha-crystallin domain. Red boxes and purple boxes indicate the I/V-X-I/V motifs and the RLFDQxFG conserved sequence, respectively. Amino acid length is indicated on the right. (B) HSPB8 schematic structure, post-translational modifications, and mutations. Post-translational modification residues are reported: phosphorylation sites are indicated in yellow, predicted myristoylation sites in orange, and a SUMOylation site in light blue. Red stars indicate HSPB8 mutations reported in the literature.
The NTD is an intrinsically disordered region enriched in Pro and Arg residues, which is highly susceptible to proteolysis [30][31]. The NTD contains the conserved RLFDQxFG motif that was shown to play an essential role in the interaction with other HSPs [32].
HSPBs can undergo post-translational modifications (Figure 1B). HSPB8 has several putative phosphorylation sites, but only Ser-14, Ser-24, Ser-27, Ser-57, Thr-63, Thr-87, and Tyr-118 phosphorylation has been described so far [33][34]. Ser-14 and Thr-63 phosphorylation was found to be mediated by protein kinase C (PKC), while p44 mitogen-activated protein kinase phosphorylates Ser-27 and Thr-87 [33][35]. Moreover, Ser-24, Ser-27, and Thr-87 are potential sites of ERK1 phosphorylation. Phosphorylation of Ser-24, Ser-27, and Thr-87 was associated with an increase in HSPB8 dimerization and a reduction in its degradation. Instead, phosphorylation of Ser-24 and Ser-27 decreases HSPB8 chaperone activity, while phosphorylation of Thr-87 increases HSPB8 chaperone activity [36]. A similar activity has been linked to Ser-57 phosphorylation mediated by cyclic AMP-dependent protein kinase (PKA) [37]. While HSPB8 phosphorylation has been evaluated through in vitro assays, its role in disease has not been elucidated.
On the other hand, HSPB8 SUMOylation at Lys-106 in MCF-7 cancer cells has been recently described. Noteworthy, HSPB8 SUMOylation is promoted by HSPB1-mediated recruitment of SUMO-2/3 enzymes. This leads to an increase in HSPB8 expression, which favors breast cancer progression (see below) [38].
Despite the fact that it is unknown whether HSPB8 is myristoylated, and that its functional significance is unknown, HSPB8 has two predicted myristoylation sites at residues Gly-62 and Gly-132 that may enhance its membrane-binding potential [39].
4. Disease-Associated Mutations of HSPB8
Few mutations have been described in the HSPB8 gene and are associated with disease (Figure 1B). The first identified mutation in the HSPB8 gene in cancer cells causes a W51C substitution in the protein sequence and was reported in one melanoma line with an important transforming potential. The mutant W51C HSPB8 acquires a predicted different secondary structure characterized by seven additional β-turns [40] and, in this way, is capable of altering HSPB8 protein–protein interaction, conferring a dominant anti-apoptotic activity to the mutated protein [40]; this greatly differs from the typical pro-apoptotic activity of wtHSPB8 in melanoma cells. Another HSPB8 anti-apoptotic mutation is the missense mutation P173H, found in human melanoma cell line MeWo. P173H HSPB8 loses its propensity to bind the transforming growth factor-beta-activated kinase 1 (TAK1), an interaction required to induce apoptosis [41], and it is thought to confer 5-Aza-deoxy-cytidine resistance to these melanoma cells.
The majority of HSPB8 mutations have been identified in patients with Charcot–Marie–Tooth (CMT) type 2L [42][43], myopathy, and distal hereditary motor neuropathy (dHMN) type IIa [44][45][46]. In particular, mutations of the conserved lysine at position 141 (K141N, K141E, K141T, and K141M), which are the most frequently described, affect HSPB8 dimerization and BAG3 interaction, causing HSPB8 toxic aggregation. Two other new HSPB8 mutations that have been identified in dHMN patients are P90L and N138T, but their role in disease needs further characterization [47]. Instead, the recently described frame shift mutation pP173Sfs*43, caused by the duplication c.515dupC in the HSPB8 gene, has been associated with autosomal dominant rimmed vacuolar myopathy. Moreover, the deletion c.508–509delCA (pQ170Gfs*45) and the duplication c.577–580dupGTCA (pT194Sfs*23) in the HSPB8 gene have been described as responsible for adult-onset axial and distal myopathy and proximal limb-girdle rimmed vacuolar myopathy, respectively [48][49]. These mutations are predicted to be translated in HSPB8 variants with an elongated C-terminal domain [46]. However, since no elongated HSPB8 has been detected in samples derived from patients, due to mRNA decay or protein instability, a haploinsufficiency mechanism has been hypothesized to explain the disease-related phenotype [46][48].
5. Modulation of HSPB8 Expression
HSPB8 is differentially expressed and displays a dual and opposite role depending upon the type of tumor considered. In fact, in some tumors, HSPB8 promotes tumor growth (e.g., in BC [77,87], lung cancer [88], multiple myeloma [89], ovarian cancer [90], and gastric cancer [91,92]), while in other tumors (e.g., in melanoma [41,93,94], leukemia [39,40,92,95], glioblastoma [69,96], hepatocarcinoma [97,98,99], and prostate cancer (PC) [40,100,101,102]). Therefore, both HSPB8 inducers and repressors could be of interest against cancer, depending upon the specific tumor to be treated.
Estrogens and selective estrogen receptor modulators (SERMs) are the first molecules identified as HSPB8 inducers [50][51][52][53][54]. Some SERMs, such as tamoxifen and 3β-diol (which is an endogenous androgenic derivative characterized by a potent estrogenic activity mediated by ERbeta [55]) induce HSPB8 expression in MCF-7 cells [50], while other SERMs, such as raloxifen and genistein, are not effective [50]. Notably, tamoxifen induces apoptosis and autophagy in BC cells [56], and it is largely used in BC treatment [57]. Unfortunately, BC cells often become resistant to tamoxifen, giving rise to more aggressive tumors. As mentioned before, HSPB8 appears to be directly involved in the acquisition of tamoxifen resistance [58][54]. HSPB8-related effects seem to be mediated by the kinase LMTK3, an upstream regulator of HSPB8 expression, that was identified as a mediator of tamoxifen resistance in BT-474 BC cells [59]. Since these adverse HSPB8 effects in tamR cells prevent patients from benefiting from tamoxifen treatment, an alternative approach has been based on AZD8055, a mTOR kinase inhibitor, which downregulates HSPB8 expression in tamR BC cells. This treatment decreases HSPB8 levels and positively correlates with reduced cell proliferation [54], indicating that AZD8055 is promising for the therapy of tamR BC tumors. Further, the anti-inflammatory statin atorvastatin (ATV) was identified as a negative modulator of HSPB8, both in in vitro and in vivo atherosclerotic models [60][61], but so far has not been tested in BC.
Not only estrogens, but also the other female steroid hormones, progestins, have been identified as HSPB8 inducers. In a gene expression profiling analysis performed on T47D BC cells treated with the synthetic progestin R5020, HSPB8 was found to be upregulated, particularly during the G2/M phase [62]. Notably, progestin-mediated HSPB8 gene transcription is regulated by a complex containing SP1, cyclin D1, and phospho-Ser345 PR, which binds with REs and SP1 DNA-binding motifs located in the promoter region of the HSPB8 gene [62].
Other potent HSPB8 inducers are several proteasome inhibitors [63], some of which are currently used as therapeutic options against cancer, due to their ability to activate autophagy [64]. In fact, when the ubiquitin-proteasome pathway is inhibited, cells upregulate autophagic proteins, including those of the CASA complex. Specifically, the proteasome inhibitors MG132, Velcade, and lactacystin upregulate HSPB8 and its co-chaperone BAG3, both at transcriptional and protein levels, in different cell types [20][65][63][66]. As mentioned above, Velcade is used for the treatment of different types of tumors, but, unfortunately, cancer cells may develop drug resistance. Similar to the case of tamoxifen, in Velcade acquired resistance, HSPB8 plays a crucial role [67]. For example, in myeloma cells, HSPB8 is robustly overexpressed in Velcade-resistant cells, where it confers a selective resistance to the drug [67].
The fact that HSPB8 expression increases when cells preferentially activate autophagy correlates with data showing that autophagy inducers may upregulate HSPB8 expression. An example is the disaccharide trehalose that activates autophagy via the lysosomal-mediated TFEB pathway [68][69]. By activating autophagy, trehalose assists the removal of several toxic misfolded proteins [68][70][71][72], and, not surprisingly, trehalose has been proven as a therapeutic approach in in vitro and in vivo cancer models [73][74][75]. Of note, still related to PQC modulation, HSPB8 and BAG3 expression has been found to respond to some proteotoxic stress via the activation of the NF-κB transcription factor [22][76]. It is known that NF-κB regulates the expression of a hundred genes, involved in inflammation, immunity, proliferation, and cell death; thus, it is difficult to foresee how and if the modulation of HSPB8 expression via NF-κB may be therapeutically useful.
Other HSPB8 inducers are the compounds able to mount the stress response mediated by the activation of HSF1, the key master regulator of HSP family gene expression. Among these are the HSPs inducer geranylgeranylacetone (GGA) [77][78] and the potent HSPs inducer multitarget small molecule N-((5-(3-(1-benzylpiperidin-4-yl)propoxy)-1-methyl-1H-indol-2-yl)methyl)-N-methylprop-2-yn-1-amine (ASS234) [79]. Both molecules have been successfully tested on AD models [80][81][82][83][84][85], exerting a protective role mediated by HSPs in general, and especially by HSPB8. Interestingly, GGA has been proven to induce apoptosis in some tumors, including melanoma [86][87], and suggested as a potential therapeutic agent for these tumors. Recalling the HSPB8 pro-apoptotic activity in melanoma cells, it would be interesting to investigate if HSPB8 may be involved in the effects observed after GGA treatment.
Using a high-throughput screening approach to find small molecules for the treatment of misfolded protein diseases, we identified two other drugs capable of positively modulating HSPB8 gene expression: colchicine and doxorubicin [24]. The two molecules both increased HSPB8 expression in a neuroblastoma ALS cell model, reducing the accumulation of toxic misfolded proteins. Colchicine is a well-known microtubule destabilizing agent, used for the treatment of gout, Mediterranean fever, Bechet’s disease, and recurrent pericarditis, and is now under investigation in a phase II clinical trial for ALS [88]. Moreover, colchicine is widely used as a research tool for the study of microtubule dynamics. Microtubules are an important target for anticancer agents and drugs that disrupt their assembly/disassembly are widely used in chemotherapy. Unfortunately, colchicine is poorly used in clinics because of its toxicity and the development of multi-drug resistance, but several drugs targeting the colchicine-binding site have recently been developed and under trials [89]. Further, the anthracycline antibiotic doxorubicin is known for its ability to kill cancer cells and commonly used to treat many forms of cancer, including BC [90]. Furthermore, in the case of doxorubicin, cancer cells may develop drug resistance [91]. Considering the negative role exerted by HSPB8 in BC cells, we can postulate that HSPB8 may play a role in acquiring resistance, and therefore this aspect in doxorubicin treatment should be carefully considered.
References
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266.
- Dokladny, K.; Myers, O.B.; Moseley, P.L. Heat shock response and autophagy—Cooperation and control. Autophagy 2015, 11, 200–213.
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111.
- Mogk, A.; Bukau, B.; Kampinga, H.H. Cellular Handling of Protein Aggregates by Disaggregation Machines. Mol. Cell 2018, 69, 214–226.
- Haslbeck, M.; Vierling, E. A first line of stress defense: Small heat shock proteins and their function in protein homeostasis. J. Mol. Biol. 2015, 427, 1537–1548.
- Boncoraglio, A.; Minoia, M.; Carra, S. The family of mammalian small heat shock proteins (HSPBs): Implications in protein deposit diseases and motor neuropathies. Int. J. Biochem. Cell Biol. 2012, 44, 1657–1669.
- Vos, M.J.; Zijlstra, M.P.; Carra, S.; Sibon, O.C.M.; Kampinga, H.H. Small heat shock proteins, protein degradation and protein aggregation diseases. Autophagy 2011, 7, 101–103.
- Kappé, G.; Franck, E.; Verschuure, P.; Boelens, W.C.; Leunissen, J.A.M.; de Jong, W.W. The human genome encodes 10 α-crystallin–related small heat shock proteins: HspB1–10. Cell Stress Chaperones 2003, 8, 53.
- De Thonel, A.; Le Mouël, A.; Mezger, V. Transcriptional regulation of small HSP—HSF1 and beyond. Int. J. Biochem. Cell Biol. 2012, 44, 1593–1612.
- Horwitz, J. Alpha-crystallin. Exp. Eye Res. 2003, 76, 145–153.
- van Montfort, R.L.; Basha, E.; Friedrich, K.L.; Slingsby, C.; Vierling, E. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat. Struct. Biol. 2001, 8, 1025–1030.
- Van Montfort, R.; Slingsby, C.; Vierling, E. Structure and function of the small heat shock protein/alpha-crystallin family of molecular chaperones. Adv. Protein Chem. 2001, 59, 105–156.
- Boelens, W.C. Structural aspects of the human small heat shock proteins related to their functional activities. Cell Stress Chaperones 2020, 25, 581–591.
- Lambert, H.; Charette, S.J.; Bernier, A.F.; Guimond, A.; Landry, J. HSP27 multimerization mediated by phosphorylation-sensitive intermolecular interactions at the amino terminus. J. Biol. Chem. 1999, 274, 9378–9385.
- Arrigo, A.-P. Human small heat shock proteins: Protein interactomes of homo- and hetero-oligomeric complexes: An update. FEBS Lett. 2013, 587, 1959–1969.
- Chowdary, T.K.; Raman, B.; Ramakrishna, T.; Rao, C.M. Mammalian Hsp22 is a heat-inducible small heat-shock protein with chaperone-like activity. Biochem. J. 2004, 381, 379–387.
- Shatov, V.M.; Strelkov, S.V.; Gusev, N.B. The Heterooligomerization of Human Small Heat Shock Proteins Is Controlled by Conserved Motif Located in the N-Terminal Domain. Int. J. Mol. Sci. 2020, 21, 4248.
- Carra, S.; Seguin, S.J.; Landry, J. HspB8 and Bag3: A new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 2008, 4, 237–239.
- Datskevich, P.N.; Mymrikov, E.V.; Gusev, N.B. Utilization of fluorescent chimeras for investigation of heterooligomeric complexes formed by human small heat shock proteins. Biochimie 2012, 94, 1794–1804.
- Crippa, V.; Sau, D.; Rusmini, P.; Boncoraglio, A.; Onesto, E.; Bolzoni, E.; Galbiati, M.; Fontana, E.; Marino, M.; Carra, S.; et al. The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum. Mol. Genet. 2010, 19, 3440–3456.
- Yu, Y.X.; Heller, A.; Liehr, T.; Smith, C.C.; Aurelian, L. Expression analysis and chromosome location of a novel gene (H11) associated with the growth of human melanoma cells. Int. J. Oncol. 2001, 18, 905–911.
- Nivon, M.; Fort, L.; Muller, P.; Richet, E.; Simon, S.; Guey, B.; Fournier, M.; Arrigo, A.-P.; Hetz, C.; Atkin, J.D.; et al. NFκB is a central regulator of protein quality control in response to protein aggregation stresses via autophagy modulation. Mol. Biol. Cell 2016, 27, 1712–1727.
- Rusmini, P.; Crippa, V.; Giorgetti, E.; Boncoraglio, A.; Cristofani, R.; Carra, S.; Poletti, A. Clearance of the mutant androgen receptor in motoneuronal models of spinal and bulbar muscular atrophy. Neurobiol. Aging 2013, 34, 2585–2603.
- Crippa, V.; D’Agostino, V.G.; Cristofani, R.; Rusmini, P.; Cicardi, M.E.; Messi, E.; Loffredo, R.; Pancher, M.; Piccolella, M.; Galbiati, M.; et al. Transcriptional induction of the heat shock protein B8 mediates the clearance of misfolded proteins responsible for motor neuron diseases. Sci. Rep. 2016, 6, 22827.
- Gamerdinger, M.; Hajieva, P.; Kaya, A.M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009, 28, 889–901.
- Carra, S.; Seguin, S.; Lambert, H.; Landry, J. HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J. Biol. Chem. 2008, 283, 1437–1444.
- Mogk, A.; Ruger-Herreros, C.; Bukau, B. Cellular Functions and Mechanisms of Action of Small Heat Shock Proteins. Ann. Rev. Microbiol. 2019, 73, 89–110.
- Saji, H.; Iizuka, R.; Yoshida, T.; Abe, T.; Kidokoro, S.-I.; Ishii, N.; Yohda, M. Role of the IXI/V motif in oligomer assembly and function of StHsp14.0, a small heat shock protein from the acidothermophilic archaeon, Sulfolobus tokodaii strain 7. Proteins 2008, 71, 771–782.
- Fuchs, M.; Poirier, D.J.; Seguin, S.J.; Lambert, H.; Carra, S.; Charette, S.J.; Landry, J. Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction. Biochem. J. 2009, 425, 245–255.
- Sudnitsyna, M.V.; Mymrikov, E.V.; Seit-Nebi, A.S.; Gusev, N.B. The role of intrinsically disordered regions in the structure and functioning of small heat shock proteins. Curr. Protein Pept. Sci. 2012, 13, 76–85.
- Aquilina, J.A.; Watt, S.J. The N-terminal domain of alphaB-crystallin is protected from proteolysis by bound substrate. Biochem. Biophys. Res. Commun. 2007, 353, 1115–1120.
- Shatov, V.M.; Weeks, S.D.; Strelkov, S.V.; Gusev, N.B. The Role of the Arginine in the Conserved N-Terminal Domain RLFDQxFG Motif of Human Small Heat Shock Proteins HspB1, HspB4, HspB5, HspB6, and HspB8. Int. J. Mol. Sci. 2018, 19, 2112.
- Benndorf, R.; Sun, X.; Gilmont, R.R.; Biederman, K.J.; Molloy, M.P.; Goodmurphy, C.W.; Cheng, H.; Andrews, P.C.; Welsh, M.J. HSP22, a new member of the small heat shock protein superfamily, interacts with mimic of phosphorylated HSP27 ((3D)HSP27). J. Biol. Chem. 2001, 276, 26753–26761.
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203.
- Molloy, M.P.; Andrews, P.C. Phosphopeptide derivatization signatures to identify serine and threonine phosphorylated peptides by mass spectrometry. Anal. Chem. 2001, 73, 5387–5394.
- Shemetov, A.A.; Seit-Nebi, A.S.; Gusev, N.B. Phosphorylation of human small heat shock protein HspB8 (Hsp22) by ERK1 protein kinase. Mol. Cell. Biochem. 2011, 355, 47–55.
- Shemetov, A.A.; Seit-Nebi, A.S.; Bukach, O.V.; Gusev, N.B. Phosphorylation by cyclic AMP-dependent protein kinase inhibits chaperone-like activity of human HSP22 in vitro. Biochemistry 2008, 73, 200–208.
- Wang, S.; Zhang, X.; Wang, H.; Wang, Y.; Chen, P.; Wang, L. Heat Shock Protein 27 Enhances SUMOylation of Heat Shock Protein B8 to Accelerate the Progression of Breast Cancer. Am. J. Pathol. 2020, 190, 2464–2477.
- Smith, C.; Yu, Y.X.; Kulka, M.; Aurelian, L. A novel human gene similar to the protein kinase (PK) coding domain of the large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10) codes for a serine-threonine PK and is expressed in melanoma cells. J. Biol. Chem. 2000, 275, 25690–25699.
- Gober, M.D.; Smith, C.C.; Ueda, K.; Toretsky, J.A.; Aurelian, L. Forced expression of the H11 heat shock protein can be regulated by DNA methylation and trigger apoptosis in human cells. J. Biol. Chem. 2003, 278, 37600–37609.
- Smith, C.; Li, B.; Liu, J.; Lee, K.-S.; Aurelian, L. The Levels of H11/HspB8 DNA methylation in human melanoma tissues and xenografts are a critical molecular marker for 5-Aza-2’-deoxycytidine therapy. Cancer Investig. 2011, 29, 383–395.
- Tang, B.; Zhao, G.; Luo, W.; Xia, K.; Cai, F.; Pan, Q.; Zhang, R.; Zhang, F.; Liu, X.; Chen, B.; et al. Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L. Hum. Genet. 2005, 116, 222–224.
- Nakhro, K.; Park, J.-M.; Kim, Y.J.; Yoon, B.R.; Yoo, J.H.; Koo, H.; Choi, B.-O.; Chung, K.W. A novel Lys141Thr mutation in small heat shock protein 22 (HSPB8) gene in Charcot-Marie-Tooth disease type 2L. Neuromuscul. Disord. 2013, 23, 656–663.
- Irobi, J.; Van Impe, K.; Seeman, P.; Jordanova, A.; Dierick, I.; Verpoorten, N.; Michalik, A.; De Vriendt, E.; Jacobs, A.; Van Gerwen, V.; et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat. Genet. 2004, 36, 597–601.
- Ghaoui, R.; Palmio, J.; Brewer, J.; Lek, M.; Needham, M.; Evilä, A.; Hackman, P.; Jonson, P.-H.; Penttilä, S.; Vihola, A.; et al. Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy. Neurology 2016, 86, 391–398.
- Al-Tahan, S.; Weiss, L.; Yu, H.; Tang, S.; Saporta, M.; Vihola, A.; Mozaffar, T.; Udd, B.; Kimonis, V. New family with HSPB8-associated autosomal dominant rimmed vacuolar myopathy. Neurol. Genet. 2019, 5, e349.
- Echaniz-Laguna, A.; Geuens, T.; Petiot, P.; Péréon, Y.; Adriaenssens, E.; Haidar, M.; Capponi, S.; Maisonobe, T.; Fournier, E.; Dubourg, O.; et al. Axonal Neuropathies due to Mutations in Small Heat Shock Proteins: Clinical, Genetic and Functional Insights into Novel Mutations. Hum. Mutat. 2017.
- Echaniz-Laguna, A.; Lornage, X.; Lannes, B.; Schneider, R.; Bierry, G.; Dondaine, N.; Boland, A.; Deleuze, J.-F.; Böhm, J.; Thompson, J.; et al. HSPB8 haploinsufficiency causes dominant adult-onset axial and distal myopathy. Acta Neuropathol. 2017, 134, 163–165.
- Nicolau, S.; Liewluck, T.; Elliott, J.L.; Engel, A.G.; Milone, M. A novel heterozygous mutation in the C-terminal region of HSPB8 leads to limb-girdle rimmed vacuolar myopathy. Neuromuscul. Disord. 2020, 30, 236–240.
- Piccolella, M.; Crippa, V.; Cristofani, R.; Rusmini, P.; Galbiati, M.; Cicardi, M.E.; Meroni, M.; Ferri, N.; Morelli, F.; Carra, S.; et al. The small heat shock protein B8 ( HSPB8 ) modulates proliferation and migration of breast cancer cells. Oncotarget 2017, 8, 10400–10415.
- Yang, C.; Trent, S.; Ionescu-Tiba, V.; Lan, L.; Shioda, T.; Sgroi, D.; Schmidt, E.V. Identification of cyclin D1- and estrogen-regulated genes contributing to breast carcinogenesis and progression. Cancer Res. 2006, 66, 11649–11658.
- Charpentier, A.H.; Bednarek, A.K.; Daniel, R.L.; Hawkins, K.A.; Laflin, K.J.; Gaddis, S.; MacLeod, M.C.; Aldaz, C.M. Effects of estrogen on global gene expression: Identification of novel targets of estrogen action. Cancer Res. 2000, 60, 5977–5983.
- Sun, X.; Fontaine, J.-M.; Bartl, I.; Behnam, B.; Welsh, M.J.; Benndorf, R. Induction of Hsp22 (HspB8) by estrogen and the metalloestrogen cadmium in estrogen receptor-positive breast cancer cells. Cell Stress Chaperones 2007, 12, 307–319.
- Shi, J.-J.; Chen, S.-M.; Guo, C.-L.; Li, Y.-X.; Ding, J.; Meng, L.-H. The mTOR inhibitor AZD8055 overcomes tamoxifen resistance in breast cancer cells by down-regulating HSPB8. Acta Pharmacol. Sin. 2018, 39, 1338–1346.
- Guerini, V.; Sau, D.; Scaccianoce, E.; Rusmini, P.; Ciana, P.; Maggi, A.; Martini, P.G.V.; Katzenellenbogen, B.S.; Martini, L.; Motta, M.; et al. The Androgen Derivative 5α-Androstane-3β,17β-Diol Inhibits Prostate Cancer Cell Migration Through Activation of the Estrogen Receptor β Subtype. Cancer Res. 2005, 65, 5445–5453.
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a molecular target for cancer treatment. Eur. J. Pharm. Sci. 2019, 134, 116–137.
- Nilsson, S.; Koehler, K.F.; Gustafsson, J.-Å. Development of subtype-selective oestrogen receptor-based therapeutics. Nat. Rev. Drug Discov. 2011, 10, 778–792.
- Gonzalez-Malerva, L.; Park, J.; Zou, L.; Hu, Y.; Moradpour, Z.; Pearlberg, J.; Sawyer, J.; Stevens, H.; Harlow, E.; LaBaer, J. High-throughput ectopic expression screen for tamoxifen resistance identifies an atypical kinase that blocks autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 2058–2063.
- Stebbing, J.; Filipovic, A.; Lit, L.C.; Blighe, K.; Grothey, A.; Xu, Y.; Miki, Y.; Chow, L.W.; Coombes, R.C.; Sasano, H.; et al. LMTK3 is implicated in endocrine resistance via multiple signaling pathways. Oncogene 2013, 32, 3371–3380.
- Chen, Q.; Xiang, J.; Gong, R.; Fang, H.; Xu, C.; Zhang, H.; Wu, Y. Atorvastatin downregulates HSP22 expression in an atherosclerotic model in vitro and in vivo. Int. J. Mol. Med. 2019, 43, 821–829.
- Tao, X.; Lu, W.; Deng, J.; Hu, Z.; Lei, Q.; Zhang, J.; Song, T.; Liu, J.; Zheng, L.; He, J. HspB8 expression in brain tissue after cerebral ischemic reperfusion and atorvastatin intervention in Sprague-Dawley rats. Neurol. Res. 2015, 37, 229–237.
- Dressing, G.E.; Knutson, T.P.; Schiewer, M.J.; Daniel, A.R.; Hagan, C.R.; Diep, C.H.; Knudsen, K.E.; Lange, C.A. Progesterone Receptor–Cyclin D1 Complexes Induce Cell Cycle–Dependent Transcriptional Programs in Breast Cancer Cells. Mol. Endocrinol. 2014, 28, 442–457.
- Cristofani, R.; Rusmini, P.; Galbiati, M.; Cicardi, M.E.; Ferrari, V.; Tedesco, B.; Casarotto, E.; Chierichetti, M.; Messi, E.; Piccolella, M.; et al. The Regulation of the Small Heat Shock Protein B8 in Misfolding Protein Diseases Causing Motoneuronal and Muscle Cell Death. Front. Neurosci. 2019, 13, 1–13.
- Richardson, P.G.; Mitsiades, C.; Hideshima, T.; Anderson, K.C. Proteasome inhibition in the treatment of cancer. Cell Cycle 2005, 4, 290–296.
- Minoia, M.; Boncoraglio, A.; Vinet, J.; Morelli, F.F.; Brunsting, J.F.; Poletti, A.; Krom, S.; Reits, E.; Kampinga, H.H.; Carra, S. BAG3 induces the sequestration of proteasomal clients into cytoplasmic puncta: Implications for a proteasome-to-autophagy switch. Autophagy 2014, 10, 1603–1621.
- Yew, E.H.J.; Cheung, N.S.; Choy, M.S.; Qi, R.Z.; Lee, A.Y.-W.; Peng, Z.F.; Melendez, A.J.; Manikandan, J.; Koay, E.S.-C.; Chiu, L.-L.; et al. Proteasome inhibition by lactacystin in primary neuronal cells induces both potentially neuroprotective and pro-apoptotic transcriptional responses: A microarray analysis. J. Neurochem. 2005, 94, 943–956.
- Hamouda, M.-A.; Belhacene, N.; Puissant, A.; Colosetti, P.; Robert, G.; Jacquel, A.; Mari, B.; Auberger, P.; Luciano, F. The small heat shock protein B8 (HSPB8) confers resistance to bortezomib by promoting autophagic removal of misfolded proteins in multiple myeloma cells. Oncotarget 2014, 5, 6252–6266.
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2019, 15, 631–651.
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338.
- Khalifeh, M.; Read, M.I.; Barreto, G.E.; Sahebkar, A. Trehalose against Alzheimer’s Disease: Insights into a Potential Therapy. Bioessays 2020, 42, e1900195.
- Mari, E.; Ricci, C.; Pieraccini, S.; Spinozzi, F.; Mariani, P.; Ortore, M.G. Trehalose Effect on the Aggregation of Model Proteins into Amyloid Fibrils. Life 2020, 10, 60.
- Khalifeh, M.; Barreto, G.E.; Sahebkar, A. Trehalose as a promising therapeutic candidate for the treatment of Parkinson’s disease. Br. J. Pharmacol. 2019, 176, 1173–1189.
- Li, Y.; Hodge, J.; Liu, Q.; Wang, J.; Wang, Y.; Evans, T.D.; Altomare, D.; Yao, Y.; Murphy, E.A.; Razani, B.; et al. TFEB is a master regulator of tumor-associated macrophages in breast cancer. J. Immunother. Cancer 2020, 8.
- Ichihara, H.; Kuwabara, K.; Matsumoto, Y. Trehalose liposomes induce apoptosis of breast tumor cells in vitro and in vivo. Biochem. Biophys. Res. Commun. 2020, 532, 505–512.
- Nikolova, B.; Antov, G.; Semkova, S.; Tsoneva, I.; Christova, N.; Nacheva, L.; Kardaleva, P.; Angelova, S.; Stoineva, I.; Ivanova, J.; et al. Bacterial Natural Disaccharide (Trehalose Tetraester): Molecular Modeling and in Vitro Study of Anticancer Activity on Breast Cancer Cells. Polymers 2020, 12, 499.
- Nivon, M.; Abou-Samra, M.; Richet, E.; Guyot, B.; Arrigo, A.-P.; Kretz-Remy, C. NF-κB regulates protein quality control after heat stress through modulation of the BAG3-HspB8 complex. J. Cell Sci. 2012, 125, 1141–1151.
- Sanbe, A.; Daicho, T.; Mizutani, R.; Endo, T.; Miyauchi, N.; Yamauchi, J.; Tanonaka, K.; Glabe, C.; Tanoue, A. Protective effect of geranylgeranylacetone via enhancement of HSPB8 induction in desmin-related cardiomyopathy. PLoS ONE 2009, 4, e5351.
- Gong, R.; Li, X.-Y.; Chen, H.-J.; Xu, C.-C.; Fang, H.-Y.; Xiang, J.; Wu, Y.-Q. Role of heat shock protein 22 in the protective effect of geranylgeranylacetone in response to oxidized-LDL. Drug Des. Dev. Ther. 2019, 13, 2619–2632.
- Ramos, E.; Romero, A.; Marco-Contelles, J.; López-Muñoz, F.; Del Pino, J. Modulation of Heat Shock Response Proteins by ASS234, Targeted for Neurodegenerative Diseases Therapy. Chem. Res. Toxicol. 2018, 31, 839–842.
- Bolea, I.; Juárez-Jiménez, J.; de los Ríos, C.; Chioua, M.; Pouplana, R.; Luque, F.J.; Unzeta, M.; Marco-Contelles, J.; Samadi, A. Synthesis, Biological Evaluation, and Molecular Modeling of Donepezil and N-[(5-(Benzyloxy)-1-methyl-1 H-indol-2-yl)methyl]-N-methylprop-2-yn-1-amine Hybrids as New Multipotent Cholinesterase/Monoamine Oxidase Inhibitors for the Treatment of Alzheimer. J. Med. Chem. 2011, 54, 8251–8270.
- Serrano, M.P.; Herrero-Labrador, R.; Futch, H.S.; Serrano, J.; Romero, A.; Fernandez, A.P.; Samadi, A.; Unzeta, M.; Marco-Contelles, J.; Martínez-Murillo, R. The proof-of-concept of ASS234: Peripherally administered ASS234 enters the central nervous system and reduces pathology in a male mouse model of Alzheimer disease. J. Psychiatry Neurosci. 2017, 42, 59–69.
- Romero, A.; Marco-Contelles, J.; Ramos, E. Highlights of ASS234: A novel and promising therapeutic agent for Alzheimer’s disease therapy. Neural Regen. Res. 2020, 15, 30–35.
- Hoshino, T.; Suzuki, K.; Matsushima, T.; Yamakawa, N.; Suzuki, T.; Mizushima, T. Suppression of Alzheimer’s disease-related phenotypes by geranylgeranylacetone in mice. PLoS ONE 2013, 8, e76306.
- Sun, Y.; Zhang, J.-R.; Chen, S. Suppression of Alzheimer’s disease-related phenotypes by the heat shock protein 70 inducer, geranylgeranylacetone, in APP/PS1 transgenic mice via the ERK/p38 MAPK signaling pathway. Exp. Ther. Med. 2017, 14, 5267–5274.
- Yokoyama, S.; Yoshinaga, T.; Matsuzaki, J.; Suzuki, H. A Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Efficacy of Teprenone in Patients with Alzheimer’s Disease. J. Alzheimers. Dis. 2019, 71, 1187–1199.
- Yoshikawa, N.; Tsuno, N.H.; Okaji, Y.; Kawai, K.; Shuno, Y.; Nagawa, H.; Oshima, N.; Takahashi, K. Isoprenoid geranylgeranylacetone inhibits human colon cancer cells through induction of apoptosis and cell cycle arrest. Anticancer Drugs 2010, 21, 850–860.
- Jo, A.R.; Jeong, H.-S.; Kim, M.-K.; Yun, H.-Y.; Baek, K.J.; Kwon, N.S.; Kim, D.-S. Geranylgeranylacetone induces apoptosis via the intrinsic pathway in human melanoma cells. Biomed. Pharmacother. 2016, 82, 15–19.
- Mandrioli, J.; Crippa, V.; Cereda, C.; Bonetto, V.; Zucchi, E.; Gessani, A.; Ceroni, M.; Chio, A.; D’Amico, R.; Monsurrò, M.R.; et al. Proteostasis and ALS: Protocol for a phase II, randomised, double-blind, placebo-controlled, multicentre clinical trial for colchicine in ALS (Co-ALS). BMJ Open 2019, 9, e028486.
- McLoughlin, E.C.; O’Boyle, N.M. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals 2020, 13, 8.
- Johnson-Arbor, K.; Dubey, R. Doxorubicin; StatPearls Publishing: Treasure Island, FL, USA, 2020.
- Al-malky, H.S.; Al Harthi, S.E.; Osman, A.-M.M. Major obstacles to doxorubicin therapy: Cardiotoxicity and drug resistance. J. Oncol. Pharm. Pract. 2020, 26, 434–444.

