 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marina Cihova | + 3656 word(s) | 3656 | 2021-03-03 07:13:09 | | | |
| 2 | Camila Xu | Meta information modification | 3656 | 2021-03-18 03:37:48 | | |
Video Upload Options
BRCA1 mutation mostly accounts for the development of TNBC with basal-like phenotype and high proliferation rate.
1. Introduction
Breast cancer represents a global burden and is considered a leading cancer type and the most common reason of cancer related deaths among the women worldwide. In 2020, it was estimated that breast cancer accounts for 30% of newly diagnosed cancer cases and 15% of cancer-related deaths in women [1]. Breast carcinoma includes several types that differ in the site at which they appear and their invasive potential. While breast carcinoma in situ refers to epithelial neoplasia that is limited to the breast duct or lobule and is considered as non-invasive or pre-invasive stage, invasive breast carcinoma describes the state when cancer cells have penetrated through the basement membrane and spread from ductolobular system to adjacent stroma. The main histological variants among the invasive breast cancers include invasive ductal and invasive lobular carcinoma with invasive ductal carcinoma being the most common type accounting for approximately 80% of invasive breast cancers [2][3]. Regarding in situ breast cancer, ductal carcinoma in situ represents the majority of diagnosed in situ breast cancers and it is considered as a precursor for development of invasive breast cancer [4]. Lobular breast carcinoma in situ is being discussed for its potential as a precursor, however, it rather indicates an increased risk for development of invasive breast cancer [5]. According to the current molecular classification, breast cancer is divided into several molecular subclasses based on gene expression profiles. The luminal A and luminal B subtypes are characterized by a gene expression pattern of luminal epithelial cells, with the luminal A subtype referring to tumors positive for estrogen receptor (ER) and/or progesterone receptor (PR) expression, negative for human epidermal growth factor receptor 2 (HER2) expression and with low levels of a proliferation marker Ki67. The luminal B subtype may show more increased expression of HER2 than the luminal A type and high Ki67 levels. HER2-enriched tumors show high HER2 expression. Basal-like breast cancer is considered as more aggressive type and is characterized by gene expression of basal epithelial genes, with tumors negative for ER and PR expression and with low HER2 expression [6]. ER-, PR- and HER2-negative tumors are referred to as triple negative breast cancer (TNBC), although these tumors include both basal-like and non-basal tumors [7]. Depending on the genetic background, the majority of breast cancer cases are sporadic with no related family cancer incidence. Sporadic breast cancer usually develops in later stages of life as a result of multiple acquired somatic mutations. However, approximately 15% of breast cancers are classified as familial with a patient´s first or second degree relative being affected [8]. Except for the genetic predisposition, familial cancers may be influenced also by non-genetic and environmental factors shared among the relatives [9]. Five to 10% of the breast cancer cases are linked with heritable germline mutations in the breast cancer related genes [10][11]. Currently, more than 25 genes have been associated with hereditary breast cancer and nearly all of them are tumor-suppressors that participate in genome stability pathways, particularly in homologous recombination DNA repair pathway and to some extent also mismatch repair, as well as inter-strand DNA crosslink repair [12]. These genes include, among others, well-described and highly penetrant tumor suppressors BRCA1 and BRCA2. Mutations in these genes were described in approximately 25% of hereditary breast cancers [13]. Estimated cumulative risk for development of breast cancer in carriers of BRCA1 and BRCA2 mutation is 72% and 69%, respectively, up to the age of 80 years [14]. BRCA1 mutation mostly accounts for the development of TNBC with basal-like phenotype and high proliferation rate [15][16][17][18]. On the contrary, carriers of BRCA2 mutation are more prone to develop ER- or PR-positive breast cancer [16]. The function of BRCA1/2 is mostly ascribed to the maintenance of genome stability through participation in DNA repair processes including homologous recombination (a key error-free DNA repair mechanism involved in double strand break repair), stabilization of DNA replication fork [19], regulation of transcription [20] and participation in DNA damage checkpoints [21] (reviewed in Nielsen et al., 2016 [12]).
BRCA1 and BRCA2 form various complexes with different proteins helping to safeguard the genome via damage signal mediation and initiation of repair by the effectors (reviewed in Savage et. al., 2015 [22], Roy et. al., 2011 [23], and summarized in Table 1).
Table 1. List of proteins with which BRCA1 and BRCA2 interact and their role in DNA repair machinery.
| Protein | Binding Partners | Function | Ref. |
|---|---|---|---|
| BRCA1 | MRN complex CtIP |
DSB end resection | [24] |
| RAP80 MERIT40 BRCC36/45 Abraxas |
stabilize/maintain DNA damage signaling and promote G2/M checkpoint arrest | [25][26] | |
| TOBP1 BACH1 (BRIP1/FANCJ) |
S-phase cell cycle arrest | [24][27] | |
| Rad51 FANCD2 |
repair replication forks stalled at DNA interstrand cross-links | [28] | |
| MLH1 ATM BLM MRN complex |
nucleotide excision repair (NER) pathway | [29] | |
| PALB2 BRCA2 |
HR-mediated DNA repair | [30] | |
| BRCA2 | BRCA1 RAD51 PALB2 |
HR-mediated DNA repair | [31][32] |
Abbreviations: MRN complex (comprised of MRE11, RAD50 and Nijmegen breakage syndrome protein 1), CtIP (CtBP-interacting protein), DSB (double-strand breaks), TOPBP1 (DNA topoisomerase 2‑binding protein 1), ATM (Ataxia-telangiectasia mutated), PALB2 (Partner and Localizer of BRCA2).
Besides the BRCA1 and BRCA2, mutations in other rare and highly penetrant genes are linked with increased risk of breast cancer development. These include STK11 (serine/threonine kinase 11; mutation is also referred to as Peutz–Jeghers syndrome) [33], PTEN (phosphatase and tensin homolog; mutation is referred to as Hamartoma-tumor syndrome) [33], TP53 (Li–Fraumeni syndrome) [34], or E-cadherin 1 (CHD1; mutation predisposes its carrier to hereditary diffuse gastric cancer with increased risk of lobular breast carcinoma) [35] and genes with moderate penetrance such as CHEK2, BRIP-1, PALB2 or ATM (reviewed in Shiovitz and Korde, 2015 [36]). Low-risk variants account for 18% of familial relative risk as identified through genome wide association study [37].
The link between BRCA1/2 mutations and high susceptibility to breast cancer development has been well-established for years. However, the potential impact of BRCA1/2 mutations on the individual cell populations within the unique tumor microenvironment (TME) and their relation to breast cancer has been understudied. The specific role of TME in all aspects of breast cancer “life cycle”, from initiation, through progression to gaining highly aggressive and metastatic phenotype is now undoubtedly one of its key hallmarks. If there is a difference between TME of sporadic and hereditary breast cancer, it has not been comprehensively addressed. Nevertheless, understanding the role of these mutations in the context of unique TME can help to better stratify the patients in terms of tumor development risk and can lead to establishment of new potential therapeutic targets by which we would be able to affect the tumor aggressiveness.
2. Breast Tumor Microenvironment: Modulator of Tumor Initiation, Progression, Metastasis and Therapy Response
The exact mechanisms responsible for cancer initiation, progression and metastasis are still not clear. The TME, composed of non-cancer cells, has been recognized as a major factor influencing the regulation of cancer cell growth, determining metastatic potential and impacting the outcome of therapy. While the stromal cells are not malignant per se, their role in influencing the tumor biology is so crucial to the survival of the tumor that these non-cancer tumor-surrounding cells have become an attractive target for therapeutic agents. Moreover, TME is now considered to be a hallmark of cancer biology [38][39][40].
The heterogeneity of TME depends on the location within the tumor. Cells forming tumor stroma, a critical component of the TME, may significantly differ at the tumor periphery and within the tumor core [41]. This is partially due to the randomly generated mutations within the tumor cells, immune cells infiltration, tumor cell necrosis and interstitial pressure [42]. Naturally, each tumor has its own unique TME, which is comprised also of non-cellular components such as extracellular matrix (ECM), soluble factors (e.g., cytokines, hormones, growth factors and enzymes) and physical properties (e.g., pH and oxygen content), which also affect properties of the TME [40]. Various cell types are involved in the bidirectional tumor-stroma interactions, mostly cancer-associated fibroblasts (CAFs), endothelial cells and pericytes, immune cells, surrounding adipocytes and mesenchymal stromal cells (MSCs), which may be resident or attracted by the tumor from distant sites [43].
CAFs are one of the most important key players in the TME. They can affect various cancer cells characteristics, such as proliferation, invasion, angiogenesis, inflammation, immunosuppression or chemoresistance. There is a remarkable heterogeneity between CAFs populations due to the expression of different surface markers indicating the different origin of the cells. They can originate from tissue resident fibroblasts, myofibroblasts, MSCs, adipocytes, pericytes, vascular smooth muscle cells, epithelial and endothelial cells (reviewed in Ting Lee et al., 2020 [44]). MSCs, which can differentiate into the CAFs in the TME, are another key part of the breast tumor stroma, as adipose tissue is one of their main sources (reviewed in Ullah et al., 2015 [45]).
Many experimental studies confirmed that MSCs have the capacity to interact with breast cancer cells (reviewed in Kucerova et al., 2011 [46]) and regulate the TME. They were shown to promote breast cancer progression and metastatic spread [47][48] as well as epithelial-to-mesenchymal transition (EMT) [49][50]. On the other hand, the reports focusing on the influence of MSCs on chemotherapy response are still somehow controversial, but they show that MSCs are also able to affect the chemoresistance of breast cancer cells [51][52][53][54][55][56]. Rapid cellular proliferation and high oxygen consumption rate results in elevated nutrient demand by the carcinoma. Tumor-associated angiogenesis, a process in which endothelial cells and pericytes build new blood vessels, occurs rapidly after the tumor formation [57]. Conversion from normal endothelial cells to the tumor-associated cells is driven by many signaling pathways modified by aberrant expression profiles. Cancer-associated endothelial cells were found to express plenty of different molecules associated with increased cancer cell survival and chemoresistance [58]. Thus, these cells that line the tumor blood vessels are important targets in cancer therapies [59].
The tumor-induced systemic changes in immune cells were also associated with cancer progression and metastasis [60]. As the metastatic process is highly inefficient, circulating tumor cells (CTC) interact for example with neutrophils by forming CTC–neutrophil clusters via vascular cell adhesion molecule 1 (VCAM-1) to expand metastatic potential [61]. It was shown that association of CTC with the complete blood count (CBC)-derived inflammation-based score (monocyte-to-lymphocyte ratio or neutrophil-to-lymphocyte ratio) may help to predict overall survival and improve the prognostication of breast cancer patients [62][63].
For further insights in tumor development and therapeutic approaches, it is important to gather more information and better understand the interplay between specific components of the TME, the associated cellular communication processes and the resulting functions of this network between cancer cells and the various tumor-associated cell populations. This might be important and shed new light in understanding germline mutation-carrying tumors, where the germline mutation affects all cells in the TME. Up to now, not much attention has been paid to how the cellular components of the TME carrying the germline mutation, such as the BRCA1/2 gene, might affect the development of breast cancer and its progression.
3. BRCA1/2-Deficient Tumor Microenvironment
The necessity to look “outside the box” of epithelial cells and search in the surrounding TME in order to elucidate BRCA1 functions and to answer questions regarding its link to more aggressive breast tumors (high expression of nuclear grade, large tumor burden, more aggressive progression and worse prognosis) has been tackled more than 13 years ago [64]. In breast cancer, it seems that heterozygous BRCA1-mutated microenvironment in germline BRCA1 mutation carriers may significantly contribute to breast cancer development by creating a pro-tumorigenic niche [65]. Several studies indicate that loss of BRCA1 in breast epithelial cells may substantially affect stromal cells residing in the TME which in turn can enhance the metastatic potential of BRCA1-deficient tumor cells [64][66][67]. This was confirmed by Plava et al. who compared in immunodeficient mouse model the co-injection of breast tumor cell line with MSCs obtained from different mammary gland sites of BRCA1 germline mutation-carrying patient [47]. MSCs from breast adipose tissue where relapsed invasive ductal carcinoma was confirmed compared to MSCs from breast adipose tissue of contralateral breast where prophylactic mastectomy was performed, increased the aggressive phenotype of the co-injected tumor cells and augmented tumor volume.
Other study showed that BRCA1-deficient breast cancer cells can transform CAFs to their altered activated phenotype, which the authors named metastasis associated fibroblasts (MAFs). MAFs can subsequently induce metastatic changes in the breast cancer cells and accompany them during metastasis [68]. It was previously shown that BRCA1 colocalizes with Ezrin, Moesin, Radixin and F-actin and controls cell motility [69]. In connection with this information, MAFs induce changes in proliferation, invasion and migration of cancer cells, which are linked to the elevated expression of EMT markers, Ezrin and CCL5 in these cells [68].
Besides the BRCA mutations per se, proteins encoded by alternative BRCA1 mRNA transcript can alter tumor microenvironment. BRCA1-IRIS, also known as IRIS (In-frame Reading of BRCA1 Intron 11 Splice variant), is an oncogene produced by the alternative splicing of the BRCA1 mRNA. Its overexpression stimulates DNA replication [70]. Therefore, IRIS expression is high in all breast cancer subtypes and even higher in TNBC. IRIS-overexpressing TNBC cells secrete interleukin 6 (IL-6) which activates STAT3, AKT, and ERK/MAPK signaling in MSCs. Thus, IRIS-overexpressing TNBC cells are able to recruit MSCs and activate them through enhancing their proliferation and migration [71].
McCullough et al. have pointed out to an intricate paracrine loop that exists between tumor and surrounding adipose stromal cells, which facilitates breast cancer progression in the mammary tissue microenvironment. Tumor cells produce factors such as IL-6 and prostaglandin E2 (PGE2) that stimulate aromatase expression in adipose stromal cells. Aromatase catalyzes estrogen production in the stromal cells, which in turn promotes estrogen-dependent growth of tumor cells. BRCA1 comes into the picture of this paracrine loop with its role in repressing aromatase gene expression in the stromal cells [64]. BRCA1 expressed in stromal cells may lead to reduced estrogen-mediated gene expression along with BRCA1-mediated repression of estrogen receptor alpha (ERα) in mammary cells and thus suppress the estrogen-dependent tumorigenesis (Figure 1a). If there is impaired BRCA1 expression in the stromal cells due to the inherited mutation, the local estrogen levels are elevated and may contribute to potential tumorigenesis by increasing genetic instability. This might seem a bit striking since BRCA1-associated tumors are largely ERα-negative. Wang et al. suggested that estrogen promotes initiation and progression of estrogen receptor negative BRCA1-deficient tumors through stimulation of cell proliferation and activation of EMT. This is dependent on protein kinase B (AKT) activation and independent of ER. Authors showed that estrogen activates the AKT pathway in BRCA1-deficient mammary tumors by enhancing the expression of p-Akt, p-mTOR, p-Gsk3 β, and p-4Ebp1, downstream targets of Akt. Along with activation of the Akt pathway, estrogen promoted EMT and proliferation in BRCA1-deficient mammary tumor cells. BRCA1-deficient tumor cells treated with Akt inhibitor AZD5363 notably inhibited estrogen-enhanced expression of p-4Ebp1, p-mTor, and p-Gsk3 β, as well as Vim (EMT marker) and p-Fra1 (EMT-inducing transcription factor) expression, suggesting that AZD5363 efficiently suppresses estrogen-enhanced Akt pathway and EMT program in BRCA1-deficient tumor cells. Interestingly, estrogen activated EMT in BRCA1-deficient, but not in BRCA1-proficient tumor cells, independent of ER [72]. To sum up, the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) pathway is stimulated by estrogen in the ER-negative BRCA1-deficient breast cancer cells and leads to enhanced tumor growth [73].
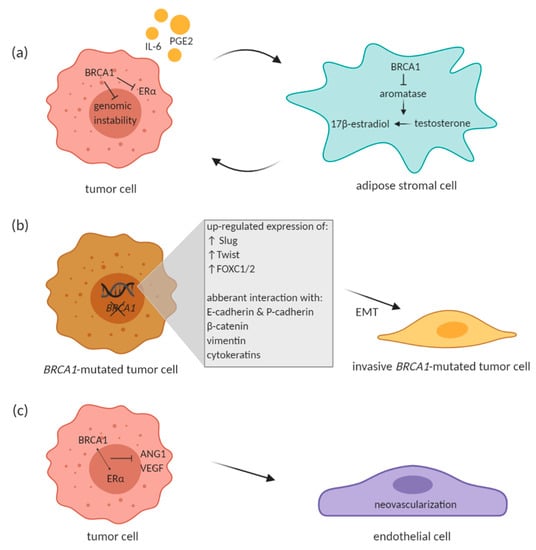
Figure 1. Proposed effect of BRCA1 on different cells in the tumor microenvironment (TME). (a) Paracrine loop between BRCA1-proficient breast tumor cell and adipose stromal cell. (b) BRCA1 mutation leads to enhanced EMT phenotype. (c) Effect of BRCA1 on endothelial cells within the TME. BRCA1 in interaction with estrogen receptor α (ERα) has been shown to inhibit VEGF and angiopoetin-1 (ANG1) transcription. Created with BioRender.com.
In ER positive breast cancer cells, the impact of estrogen on EMT and the ability of ERα signaling to crosstalk with EMT regulators such as Snail and Slug has been suggested and described few years ago [74]. In a study by Bouris et al., the role of estrogen/ERα signaling in EMT in breast cancer cells was documented in an experiment, in which the ERα in MCF-7 cells was knocked-down through specific shRNA lentiviral particles. The cells changed phenotypically along with significant change in gene and protein expression of several markers typical for EMT, such as complete loss of E-cadherin, significantly induced expression of vimentin and fibronectin (mesenchymal protein markers) as well as of ΕΜΤ-related transcriptional regulators, such as ΖΕB1/δEF1 and SNAIL2/SLUG. ERα suppression also lead to altered expression of EGFR and HER2 receptor tyrosine kinases, and various extracellular matrix metalloproteinases and components of the plasminogen activation system. Compared to wild type MCF-7, the ERα-silenced MCF-7 cells exhibited enhanced proliferation, migration and invasion [75].
3.1. EMT Process in BRCA-Deficient Tumors
The EMT is a complex cellular process involved in embryogenesis, tissue reparation and wound healing, and also in tumorigenesis. During EMT, epithelial cells acquire mesenchymal phenotype and migratory and invasive properties [76][77][78]. There are plethora of biological events and signaling pathways involved. Regarding the carcinogenesis, changes in EMT regulatory pathways lead to more aggressive cellular phenotype—changes in the polarity of the cell, loss of cellular adhesions, detachment, migration, intravasation and ability to survive in the vascular system, extravasation, and metastasis. Therefore, EMT events are linked with progression from pre-invasive to the invasive state of cancer and metastatic disease [78]. To initiate all of that, many biological processes and molecules are involved, including the TGF-β, Notch and Wnt pathways, effects of the TME such as hypoxia and expression of different microRNAs [79]. Several transcription factors, including Snail 1/2 (also referred to as Snail and Slug), ZEB 1/2, Ets-1, FOXC1/2 and Twist affect expression of many genes that were identified as initiators of EMT [80][81].
In breast cancer, EMT has the greatest impact on basal-like tumors. Many studies showed that basal-like breast tumors represent the most aggressive and deadly breast cancer subtype with high metastatic ability [76][82]. As mentioned earlier, germline mutations in the tumor suppressor gene BRCA1 increase the risk of developing basal-like breast tumors with high metastasis and poor prognosis [15][16][17][18][83][84]. It was shown that aberrant luminal stem cells can more likely give origin to basal-like tumors than basal progenitors [85][86][87]. Bai et al. showed that a germline mutation or mammary epithelia-specific deletion of BRCA1 is responsible for the activation of EMT transcription factors and thus induction of EMT, dedifferentiation of luminal stem cells and expansion of basal and cancer stem cells. These events are important in development of basal-like tumors [81].
Many other studies reported the relationship between BRCA1 mutation and EMT transcription factors. In BRCA1-mutated tumors, predominantly basal-like tumors, Slug was reported to be up-regulated although BRCA1 is not a transcriptional repressor [88][89]. Also Twist and FOXC1/2 are over-expressed in those types of tumors with BRCA1 mutation, because BRCA1 acts like a direct repressor of these transcriptional factors under normal conditions [81][90]. In addition to the interaction with transcription factors, presence of BRCA1 mutation was correlated with regulation of EMT through cell surface proteins E- and P-cadherin, cytoskeletal protein such as β -catenin, vimentin and cytokeratins (Figure 1b) (reviewed in Sengodan et al., 2018 [80]). As mentioned above, BRCA1 was also described to reduce breast cancer cell migration through ubiquitination of ezrin–radixin–moexin protein complex that is important in regulation of cellular motility and spreading [69].
The role of EMT in BRCA2-deficient tumors was correlated with primary resistance or relapse after PARP inhibitor treatment associated with rapid expression of EMT-associated markers in a treatment-dependent manner [91]. EMT-like phenotype correlated with a high expression of the ABCB1B gene and was associated with multidrug resistance also in BRCA2-deficient sarcomatoid mammary tumors. ABCB1B gene encodes for drug efflux transporter P-glycoprotein and its inhibition partially re-sensitized sarcomatoid tumors to the PARP inhibitors [92].
3.2. Impact of BRCA Deficiency on Tumor Neovascularization
Studies have shown that intricate interaction between BRCA1-deficient tumor cells and the surrounding stroma may also be manifested in promoting endothelial cell survival and vascularization. Tumor growth and progression is accompanied by development of vascular network within the tumor. Formation of new vasculature from existing vascular network, a process called angiogenesis, is one of the hallmarks of growing tumors and its onset is also known as angiogenic switch. Angiogenesis is under the control of number of pro- and anti-angiogenic factors like angiogenic vascular endothelial growth factor (VEGF), angiopoietins (Ang) or basic fibroblast growth factor (bFGF). Anti-angiogenic factors include for example thrombospondins or circulating endostatin and angiostatin (reviewed in Huang and Bao, 2004 [93]).
Extensive cancer cell proliferation demands higher oxygen utilization and together with inadequate blood supply within the tumor results in formation of hypoxia which is often observed in many tumors. Hypoxic conditions drive increase in activity of hypoxia inducible factors (HIF) [94]. HIF1α (a subunit of HIF1 heterodimer) is under normoxic conditions ubiquitinated and degraded by proteasome, although its expression is in cancer cells elevated during hypoxia [95]. Accumulated HIF1α dimerizes with HIF1β subunit and, together with its co-activator proteins, HIF1 participates in activation of its target genes [94]. Among them, VEGF has been widely described as a key component of angiogenic regulation and blood vessel formation [96]. VEGF, together with HIF1α, are also considered as important factors involved in cancer progression and dissemination [97][98].
Increase in VEGF and HIF1α expression has been observed in BRCA1/2-related and hereditary breast cancer when compared to sporadic breast cancer as well as in BRCA1/2-related hereditary breast cancer compared to other types of hereditary breast cancer [99]. Moreover, BRCA1 in interaction with ERα has been shown to inhibit VEGF transcription and protein expression through estrogen signaling pathway (Figure 1c). BRCA1 in interaction with ERα prevented VEGF expression by its binding to VEGF promoter and thus suppressed its activity [100]. Elevated expression of Ang-1, Ang-2 and VEGF was also described in BRCA1/2-mutated tumors of hereditary breast cancer patients [101]. Similarly, mammary tumors from BRCA1-deficient mice exhibited increased expression of Ang-1 together with notable vascular growth [102]. In this study, BRCA1 was also described to inhibit Ang-1 transcription by forming a repressive complex with CtIP (CtBP-interacting protein) and ZBRK1 (Zinc finger and BRCA1-interacting protein with KRAB domain 1). This complex then represses Ang-1 through ZBRK1 recognition site on Ang-1 promoter [102]. Moreover, Danza et al. highlighted involvement of miRNA-mediated angiogenesis in BRCA1/2-deficient tumors. The authors showed miR-578 and miR-573 involvement in BRCA1/2-related angiogenesis by affecting VEGF, FAK and HIF-1 signaling pathways [103]. The above-mentioned studies therefore suggest an involvement of BRCA1/2 in tumor angiogenesis and cancer progression.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30.
- Zengel, B.; Yararbas, U.; Duran, A.; Uslu, A.; Eliyatkin, N.; Demirkiran, M.A.; Cengiz, F.; Simsek, C.; Postaci, H.; Vardar, E.; et al. Comparison of the clinicopathological features of invasive ductal, invasive lobular, and mixed (invasive ductal + invasive lobular) carcinoma of the breast. Breast Cancer 2015, 22, 374–381.
- Bharat, A.; Gao, F.; Margenthaler, J.A. Tumor characteristics and patient outcomes are similar between invasive lobular and mixed invasive ductal/lobular breast cancers but differ from pure invasive ductal breast cancers. Am. J. Surg. 2009, 198, 516–519.
- Ward, E.M.; DeSantis, C.E.; Lin, C.C.; Kramer, J.L.; Jemal, A.; Kohler, B.; Brawley, O.W.; Gansler, T. Cancer statistics: Breast cancer in situ. CA Cancer J. Clin. 2015, 65, 481–495.
- Chuba, P.J.; Hamre, M.R.; Yap, J.; Severson, R.K.; Lucas, D.; Shamsa, F.; Aref, A. Bilateral risk for subsequent breast cancer after lobular carcinoma-in-situ: Analysis of surveillance, epidemiology, and end results data. J. Clin. Oncol. 2005, 23, 5534–5541.
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752.
- Bertucci, F.; Finetti, P.; Cervera, N.; Esterni, B.; Hermitte, F.; Viens, P.; Birnbaum, D. How basal are triple-negative breast cancers? Int. J. Cancer 2008, 123, 236–240.
- Zheng, G.; Yu, H.; Hemminki, A.; Forsti, A.; Sundquist, K.; Hemminki, K. Familial associations of female breast cancer with other cancers. Int. J. Cancer 2017, 141, 2253–2259.
- Couto, E.; Hemminki, K. Estimates of heritable and environmental components of familial breast cancer using family history information. Br. J. Cancer 2007, 96, 1740–1742.
- Claus, E.B.; Schildkraut, J.M.; Thompson, W.D.; Risch, N.J. The genetic attributable risk of breast and ovarian cancer. Cancer 1996, 77, 2318–2324.
- Apostolou, P.; Fostira, F. Hereditary breast cancer: The era of new susceptibility genes. Biomed. Res. Int 2013, 2013, 747318.
- Nielsen, F.C.; van Overeem Hansen, T.; Sorensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612.
- Kast, K.; Rhiem, K.; Wappenschmidt, B.; Hahnen, E.; Hauke, J.; Bluemcke, B.; Zarghooni, V.; Herold, N.; Ditsch, N.; Kiechle, M.; et al. Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J. Med. Genet. 2016, 53, 465–471.
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416.
- Lakhani, S.R.; Reis-Filho, J.S.; Fulford, L.; Penault-Llorca, F.; van der Vijver, M.; Parry, S.; Bishop, T.; Benitez, J.; Rivas, C.; Bignon, Y.J.; et al. Prediction of BRCA1 status in patients with breast cancer using estrogen receptor and basal phenotype. Clin. Cancer Res. 2005, 11, 5175–5180.
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 2012, 21, 134–147.
- Foulkes, W.D.; Stefansson, I.M.; Chappuis, P.O.; Begin, L.R.; Goffin, J.R.; Wong, N.; Trudel, M.; Akslen, L.A. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J. Natl. Cancer Inst. 2003, 95, 1482–1485.
- Armes, J.E.; Trute, L.; White, D.; Southey, M.C.; Hammet, F.; Tesoriero, A.; Hutchins, A.M.; Dite, G.S.; McCredie, M.R.; Giles, G.G.; et al. Distinct molecular pathogeneses of early-onset breast cancers in BRCA1 and BRCA2 mutation carriers: A population-based study. Cancer Res. 1999, 59, 2011–2017.
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542.
- Gardini, A.; Baillat, D.; Cesaroni, M.; Shiekhattar, R. Genome-wide analysis reveals a role for BRCA1 and PALB2 in transcriptional co-activation. EMBO J. 2014, 33, 890–905.
- Fabbro, M.; Savage, K.; Hobson, K.; Deans, A.J.; Powell, S.N.; McArthur, G.A.; Khanna, K.K. BRCA1-BARD1 complexes are required for p53Ser-15 phosphorylation and a G1/S arrest following ionizing radiation-induced DNA damage. J. Biol. Chem. 2004, 279, 31251–31258.
- Savage, K.I.; Harkin, D.P. BRCA1, a ‘complex’ protein involved in the maintenance of genomic stability. FEBS J. 2015, 282, 630–646.
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78.
- Greenberg, R.A.; Sobhian, B.; Pathania, S.; Cantor, S.B.; Nakatani, Y.; Livingston, D.M. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006, 20, 34–46.
- Coleman, K.A.; Greenberg, R.A. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J. Biol. Chem. 2011, 286, 13669–13680.
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198.
- Xie, J.; Peng, M.; Guillemette, S.; Quan, S.; Maniatis, S.; Wu, Y.; Venkatesh, A.; Shaffer, S.A.; Brosh, R.M., Jr.; Cantor, S.B. FANCJ/BACH1 acetylation at lysine 1249 regulates the DNA damage response. PLoS Genet. 2012, 8, e1002786.
- Kim, H.; D’Andrea, A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012, 26, 1393–1408.
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000, 14, 927–939.
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol. 2009, 19, 524–529.
- Liu, C.; Srihari, S.; Cao, K.A.; Chenevix-Trench, G.; Simpson, P.T.; Ragan, M.A.; Khanna, K.K. A fine-scale dissection of the DNA double-strand break repair machinery and its implications for breast cancer therapy. Nucleic Acids Res. 2014, 42, 6106–6127.
- Christou, C.M.; Kyriacou, K. BRCA1 and Its Network of Interacting Partners. Biology 2013, 2, 40–63.
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407.
- Mai, P.L.; Best, A.F.; Peters, J.A.; DeCastro, R.M.; Khincha, P.P.; Loud, J.T.; Bremer, R.C.; Rosenberg, P.S.; Savage, S.A. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 2016, 122, 3673–3681.
- Pharoah, P.D.; Guilford, P.; Caldas, C. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001, 121, 1348–1353.
- Shiovitz, S.; Korde, L.A. Genetics of breast cancer: A topic in evolution. Ann. Oncol. 2015, 26, 1291–1299.
- Michailidou, K.; Lindstrom, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemacon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94.
- Place, A.E.; Jin Huh, S.; Polyak, K. The microenvironment in breast cancer progression: Biology and implications for treatment. Breast Cancer Res. 2011, 13, 227.
- Plava, J.; Cihova, M.; Burikova, M.; Matuskova, M.; Kucerova, L.; Miklikova, S. Recent advances in understanding tumor stroma-mediated chemoresistance in breast cancer. Mol. Cancer 2019, 18, 67.
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152.
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016, 29, 587–601.
- Orlando, P.A.; Gatenby, R.A.; Brown, J.S. Tumor evolution in space: The effects of competition colonization tradeoffs on tumor invasion dynamics. Front. Oncol. 2013, 3, 45.
- Eiro, N.; Gonzalez, L.O.; Fraile, M.; Cid, S.; Schneider, J.; Vizoso, F.J. Breast Cancer Tumor Stroma: Cellular Components, Phenotypic Heterogeneity, Intercellular Communication, Prognostic Implications and Therapeutic Opportunities. Cancers 2019, 11, 664.
- Lee, Y.T.; Tan, Y.J.; Falasca, M.; Oon, C.E. Cancer-Associated Fibroblasts: Epigenetic Regulation and Therapeutic Intervention in Breast Cancer. Cancers 2020, 12, 2949.
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells—Current trends and future prospective. Biosci. Rep. 2015, 35.
- Kucerova, L.; Kovacovicova, M.; Polak, S.; Bohac, M.; Fedeles, J.; Palencar, D.; Matuskova, M. Interaction of human adipose tissue-derived mesenchymal stromal cells with breast cancer cells. Neoplasma 2011, 58, 361–370.
- Plava, J.; Cihova, M.; Burikova, M.; Bohac, M.; Adamkov, M.; Drahosova, S.; Rusnakova, D.; Pindak, D.; Karaba, M.; Simo, J.; et al. Permanent Pro-Tumorigenic Shift in Adipose Tissue-Derived Mesenchymal Stromal Cells Induced by Breast Malignancy. Cells 2020, 9, 480.
- Yu, P.F.; Huang, Y.; Xu, C.L.; Lin, L.Y.; Han, Y.Y.; Sun, W.H.; Hu, G.H.; Rabson, A.B.; Wang, Y.; Shi, Y.F. Downregulation of CXCL12 in mesenchymal stromal cells by TGFbeta promotes breast cancer metastasis. Oncogene 2017, 36, 840–849.
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563.
- Martin, F.T.; Dwyer, R.M.; Kelly, J.; Khan, S.; Murphy, J.M.; Curran, C.; Miller, N.; Hennessy, E.; Dockery, P.; Barry, F.P.; et al. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: Stimulation of epithelial to mesenchymal transition (EMT). Breast Cancer Res. Treat. 2010, 124, 317–326.
- Shi, Z.; Yang, W.M.; Chen, L.P.; Yang, D.H.; Zhou, Q.; Zhu, J.; Chen, J.J.; Huang, R.C.; Chen, Z.S.; Huang, R.P. Enhanced chemosensitization in multidrug-resistant human breast cancer cells by inhibition of IL-6 and IL-8 production. Breast Cancer Res. Treat. 2012, 135, 737–747.
- Chen, D.R.; Lu, D.Y.; Lin, H.Y.; Yeh, W.L. Mesenchymal stem cell-induced doxorubicin resistance in triple negative breast cancer. Biomed. Res. Int 2014, 2014, 532161.
- Yeh, W.L.; Tsai, C.F.; Chen, D.R. Peri-foci adipose-derived stem cells promote chemoresistance in breast cancer. Stem. Cell Res. Ther. 2017, 8, 177.
- Roodhart, J.M.; Daenen, L.G.; Stigter, E.C.; Prins, H.J.; Gerrits, J.; Houthuijzen, J.M.; Gerritsen, M.G.; Schipper, H.S.; Backer, M.J.; van Amersfoort, M.; et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell 2011, 20, 370–383.
- Kucerova, L.; Skolekova, S.; Matuskova, M.; Bohac, M.; Kozovska, Z. Altered features and increased chemosensitivity of human breast cancer cells mediated by adipose tissue-derived mesenchymal stromal cells. BMC Cancer 2013, 13, 535.
- Skolekova, S.; Matuskova, M.; Bohac, M.; Toro, L.; Durinikova, E.; Tyciakova, S.; Demkova, L.; Gursky, J.; Kucerova, L. Erratum to: Cisplatin-induced mesenchymal stromal cells-mediated mechanism contributing to decreased antitumor effect in breast cancer cells. Cell Commun. Signal. 2016, 14, 7.
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322.
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84.
- Hida, K.; Maishi, N.; Annan, D.A.; Hida, Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 1272.
- Wang, L.; Simons, D.L.; Lu, X.; Tu, T.Y.; Avalos, C.; Chang, A.Y.; Dirbas, F.M.; Yim, J.H.; Waisman, J.; Lee, P.P. Breast cancer induces systemic immune changes on cytokine signaling in peripheral blood monocytes and lymphocytes. EBioMedicine 2020, 52, 102631.
- Saini, M.; Szczerba, B.M.; Aceto, N. Circulating Tumor Cell-Neutrophil Tango along the Metastatic Process. Cancer Res. 2019, 79, 6067–6073.
- Miklikova, S.; Minarik, G.; Sedlackova, T.; Plava, J.; Cihova, M.; Jurisova, S.; Kalavska, K.; Karaba, M.; Benca, J.; Smolkova, B.; et al. Inflammation-Based Scores Increase the Prognostic Value of Circulating Tumor Cells in Primary Breast Cancer. Cancers 2020, 12, 1134.
- De Giorgi, U.; Mego, M.; Scarpi, E.; Giordano, A.; Giuliano, M.; Valero, V.; Alvarez, R.H.; Ueno, N.T.; Cristofanilli, M.; Reuben, J.M. Association between circulating tumor cells and peripheral blood monocytes in metastatic breast cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919866065.
- McCullough, S.D.; Hu, Y.; Li, R. BRCA1 in initiation, invasion, and metastasis of breast cancer: A perspective from the tumor microenvironment. In Metastasis of Breast Cancer; Mansel, R.E., Fodstad, O., Jiang, W.G., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 31–46.
- Li, C.M.; Oren, Y.; Regev, A.; Brugge, J.S. Abstract PR06: Contribution of mutant microenvironment to hereditary cancer: Single-cell gene expression profiling of a genetically engineered mouse model of human hereditary BRCA1-related breast cancer. Cancer Res. 2018, 78, PR06.
- Ghosh, S.; Lu, Y.; Katz, A.; Hu, Y.; Li, R. Tumor suppressor BRCA1 inhibits a breast cancer-associated promoter of the aromatase gene (CYP19) in human adipose stromal cells. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E246–E252.
- Weber, F.; Shen, L.; Fukino, K.; Patocs, A.; Mutter, G.L.; Caldes, T.; Eng, C. Total-genome analysis of BRCA1/2-related invasive carcinomas of the breast identifies tumor stroma as potential landscaper for neoplastic initiation. Am. J. Hum. Genet. 2006, 78, 961–972.
- Hemalatha, S.K.; Sengodan, S.K.; Nadhan, R.; Dev, J.; Sushama, R.R.; Somasundaram, V.; Thankappan, R.; Rajan, A.; Latha, N.R.; Varghese, G.R.; et al. Brcal Defective Breast Cancer Cells Induce in vitro Transformation of Cancer Associated Fibroblasts (CAFs) to Metastasis Associated Fibroblasts (MAF). Sci. Rep. 2018, 8, 13903.
- Coene, E.D.; Gadelha, C.; White, N.; Malhas, A.; Thomas, B.; Shaw, M.; Vaux, D.J. A novel role for BRCA1 in regulating breast cancer cell spreading and motility. J. Cell Biol. 2011, 192, 497–512.
- ElShamy, W.M.; Livingston, D.M. Identification of BRCA1-IRIS, a BRCA1 locus product. Nat. Cell Biol. 2004, 6, 954–967.
- Ryan, D.; Paul, B.T.; Koziol, J.; ElShamy, W.M. The pro- and anti-tumor roles of mesenchymal stem cells toward BRCA1-IRIS-overexpressing TNBC cells. Breast Cancer Res. 2019, 21, 53.
- Wang, C.; Bai, F.; Zhang, L.H.; Scott, A.; Li, E.; Pei, X.H. Estrogen promotes estrogen receptor negative BRCA1-deficient tumor initiation and progression. Breast Cancer Res. 2018, 20, 74.
- Gorrini, C.; Gang, B.P.; Bassi, C.; Wakeham, A.; Baniasadi, S.P.; Hao, Z.; Li, W.Y.; Cescon, D.W.; Li, Y.T.; Molyneux, S.; et al. Estrogen controls the survival of BRCA1-deficient cells via a PI3K-NRF2-regulated pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 4472–4477.
- Saha Roy, S.; Vadlamudi, R.K. Role of estrogen receptor signaling in breast cancer metastasis. Int. J. Breast Cancer 2012, 2012, 654698.
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632.
- Micalizzi, D.S.; Ford, H.L. Epithelial-mesenchymal transition in development and cancer. Future Oncol. 2009, 5, 1129–1143.
- Fedele, M.; Cerchia, L.; Chiappetta, G. The Epithelial-to-Mesenchymal Transition in Breast Cancer: Focus on Basal-Like Carcinomas. Cancers 2017, 9, 134.
- Felipe Lima, J.; Nofech-Mozes, S.; Bayani, J.; Bartlett, J.M. EMT in Breast Carcinoma-A Review. J. Clin. Med. 2016, 5.
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273.
- Sengodan, S.K.; Sreelatha, K.H.; Nadhan, R.; Srinivas, P. Regulation of epithelial to mesenchymal transition by BRCA1 in breast cancer. Crit. Rev. Oncol. Hematol. 2018, 123, 74–82.
- Bai, F.; Chan, H.L.; Scott, A.; Smith, M.D.; Fan, C.; Herschkowitz, J.I.; Perou, C.M.; Livingstone, A.S.; Robbins, D.J.; Capobianco, A.J.; et al. BRCA1 suppresses epithelial-to-mesenchymal transition and stem cell dedifferentiation during mammary and tumor development. Cancer Res. 2014, 74, 6161–6172.
- Scimeca, M.; Antonacci, C.; Colombo, D.; Bonfiglio, R.; Buonomo, O.C.; Bonanno, E. Emerging prognostic markers related to mesenchymal characteristics of poorly differentiated breast cancers. Tumour Biol. 2016, 37, 5427–5435.
- Zhu, Y.; Wu, J.; Zhang, C.; Sun, S.; Zhang, J.; Liu, W.; Huang, J.; Zhang, Z. BRCA mutations and survival in breast cancer: An updated systematic review and meta-analysis. Oncotarget 2016, 7, 70113–70127.
- Song, Y.; Barry, W.T.; Seah, D.S.; Tung, N.M.; Garber, J.E.; Lin, N.U. Patterns of recurrence and metastasis in BRCA1/BRCA2-associated breast cancers. Cancer 2020, 126, 271–280.
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009, 15, 907–913.
- Molyneux, G.; Geyer, F.C.; Magnay, F.A.; McCarthy, A.; Kendrick, H.; Natrajan, R.; Mackay, A.; Grigoriadis, A.; Tutt, A.; Ashworth, A.; et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem. Cell 2010, 7, 403–417.
- Bai, F.; Smith, M.D.; Chan, H.L.; Pei, X.H. Germline mutation of Brca1 alters the fate of mammary luminal cells and causes luminal-to-basal mammary tumor transformation. Oncogene 2013, 32, 2715–2725.
- Lindeman, G.J.; Visvader, J.E. Cell fate takes a slug in BRCA1-associated breast cancer. Breast Cancer Res. 2011, 13, 306.
- Proia, T.A.; Keller, P.J.; Gupta, P.B.; Klebba, I.; Jones, A.D.; Sedic, M.; Gilmore, H.; Tung, N.; Naber, S.P.; Schnitt, S.; et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem. Cell 2011, 8, 149–163.
- Mani, S.A.; Yang, J.; Brooks, M.; Schwaninger, G.; Zhou, A.; Miura, N.; Kutok, J.L.; Hartwell, K.; Richardson, A.L.; Weinberg, R.A. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2007, 104, 10069–10074.
- Ordonez, L.D.; Hay, T.; McEwen, R.; Polanska, U.M.; Hughes, A.; Delpuech, O.; Cadogan, E.; Powell, S.; Dry, J.; Tornillo, G.; et al. Rapid activation of epithelial-mesenchymal transition drives PARP inhibitor resistance in Brca2-mutant mammary tumours. Oncotarget 2019, 10, 2586–2606.
- Jaspers, J.E.; Sol, W.; Kersbergen, A.; Schlicker, A.; Guyader, C.; Xu, G.; Wessels, L.; Borst, P.; Jonkers, J.; Rottenberg, S. BRCA2-deficient sarcomatoid mammary tumors exhibit multidrug resistance. Cancer Res. 2015, 75, 732–741.
- Huang, Z.; Bao, S.D. Roles of main pro- and anti-angiogenic factors in tumor angiogenesis. World J. Gastroenterol. 2004, 10, 463–470.
- Gilkes, D.M.; Semenza, G.L. Role of hypoxia-inducible factors in breast cancer metastasis. Future Oncol. 2013, 9, 1623–1636.
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997, 272, 22642–22647.
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69 (Suppl. 3), 4–10.
- Liao, D.; Corle, C.; Seagroves, T.N.; Johnson, R.S. Hypoxia-inducible factor-1alpha is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res. 2007, 67, 563–572.
- Masoumi Moghaddam, S.; Amini, A.; Morris, D.L.; Pourgholami, M.H. Significance of vascular endothelial growth factor in growth and peritoneal dissemination of ovarian cancer. Cancer Metastasis Rev. 2012, 31, 143–162.
- Saponaro, C.; Malfettone, A.; Ranieri, G.; Danza, K.; Simone, G.; Paradiso, A.; Mangia, A. VEGF, HIF-1alpha expression and MVD as an angiogenic network in familial breast cancer. PLoS ONE 2013, 8, e53070.
- Kawai, H.; Li, H.; Chun, P.; Avraham, S.; Avraham, H.K. Direct interaction between BRCA1 and the estrogen receptor regulates vascular endothelial growth factor (VEGF) transcription and secretion in breast cancer cells. Oncogene 2002, 21, 7730–7739.
- Danza, K.; Pilato, B.; Lacalamita, R.; Addati, T.; Giotta, F.; Bruno, A.; Paradiso, A.; Tommasi, S. Angiogenetic axis angiopoietins/Tie2 and VEGF in familial breast cancer. Eur. J. Hum. Genet. 2013, 21, 824–830.
- Furuta, S.; Wang, J.M.; Wei, S.; Jeng, Y.M.; Jiang, X.; Gu, B.; Chen, P.L.; Lee, E.Y.; Lee, W.H. Removal of BRCA1/CtIP/ZBRK1 repressor complex on ANG1 promoter leads to accelerated mammary tumor growth contributed by prominent vasculature. Cancer Cell 2006, 10, 13–24.
- Danza, K.; De Summa, S.; Pinto, R.; Pilato, B.; Palumbo, O.; Merla, G.; Simone, G.; Tommasi, S. MiR-578 and miR-573 as potential players in BRCA-related breast cancer angiogenesis. Oncotarget 2015, 6, 471–483.

