 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yutaka Furutani | + 3127 word(s) | 3127 | 2021-03-08 02:55:57 | | | |
| 2 | Peter Tang | Meta information modification | 3127 | 2021-03-09 05:19:57 | | |
Video Upload Options
Transglutaminase 2 (TG2) is a crosslinking enzyme that forms a covalent bond between lysine and glutamine. TG2 plays important roles in diverse cellular processes, including extracellular matrix stabilization, cytoskeletal function, cell motility, adhesion, signal transduction, apoptosis, and cell survival.
1. Introduction
Sepsis results in lethal organ dysfunction, which is increasing in prevalence due to the dysregulated host response to infection [1]. It is a global health crisis, affecting over 50 million people worldwide and causing approximately 5.3 million deaths annually. According to a systematic review, it is recognized as the major cause of admission to the intensive care unit (ICU) and death worldwide [2][3][4]. In 2017, the World Health Organization (WHO) suggested that sepsis should be recognized as a global health priority, and its prevention, diagnosis, and management should be improved [3][4][5]. Moreover, sepsis is frequently found to be the cause of death due to infections worldwide. Early research predominantly focused on bacterial infection, which underlies the etiology of 80% of adult septic patients [6][7]. In 2020, with the spread of SARS-CoV-2, high mortality, and stressed ICU capacity, research on sepsis turned toward the host response to the invading virus. It has been reported that approximately 2–5% for patients with COVID-19 suffer multi-organ injury after 8–10 days, which is typical of sepsis [8][9][10]. Despite this alarming situation, sepsis, which is little known to the general public, remains relatively neglected.
Billions of dollars have been invested by the government and the pharmaceutical industry in the past 40 years for the development of specific immune system-targeting adjunct therapies for sepsis. However, to date, none of the numerous clinical trials using a single adjuvant treatment has achieved a breakthrough [11][12][13]. Basic and clinical scientists agree that rather than these drugs being ineffective, this observation could be due to the inability to perform continuous immunomonitoring for each patient who may suffer from different conditions and extent of sepsis [14]. Therefore, ongoing research aims to utilize individualized management strategies that are matched to personal clinical profiles [15].
Transglutaminase 2 (TG2) is a multifunctional enzyme that is associated with a variety of physiological and pathological functions, including cell growth, differentiation and development, cell adhesion and morphology, cytoskeletal rearrangement, extracellular matrix stabilization, inflammatory processes, receptor-mediated endocytosis, and apoptosis [16][17]. TG2 is a calcium-dependent transamidase that catalyzes the crosslinking of glutamine to lysine residues within or between proteins [18]. Given these functions, TG2 plays critical roles in the pathogenesis of a variety of human diseases, including inflammatory diseases, such as colitis, celiac disease, rheumatoid arthritis, cardiovascular disease, neurodegenerative diseases, fibrosis, cancer, and sepsis [19][20][21][22][23].
2. Physiological and Pathological Role of TG in Inflammation
TG is an enzyme that crosslinks lysine and glutamine within or between proteins by forming an isopeptide bond. TG is a family of enzymes consisting of TG1–7 and Factor XIIIa (FXIIIa). TG1, TG3, and TG5 are mainly expressed in the skin and esophagus, where they stabilize the cornified cell envelope by crosslinking the terminally differentiated keratinocytes [24]. TG1 is essential for the assembly and organization of the barrier structures to form a cornified envelope [25]. TG1-deficient mice show the ichthyosiform skin phenotype and develop massive hyperkeratosis [26]. TG3-deficient mouse skin is more responsive to the action of the pro-inflammatory drug imiquimod than WT mouse skin, resulting in psoriasis [27]. TG5 contributes to the hyperkeratotic phenotype in ichthyosis and psoriasis [28]. TG2 is ubiquitously expressed and is regulated by Ca2+ and guanosine triphosphate (GTP). TG4 is specifically expressed in the prostate and crosslinks proteins in the seminal vesicle fluid [29]. FXIIIa is a blood coagulation factor that forms fibrin-based clots by crosslinking [30].
The function of TG2 has been extensively studied and is known to be regulated by Ca2+ and GTP. TG2 is composed of 667 amino acids and has a catalytic core domain with an activity center similar to cysteine proteases. The GTP-bound TG2 exists in a closed form and its conformation changes to the open form upon Ca2+-binding, which also activates transamidase activity (Figure 1). Its transamidation activity can be blocked by using Ca2+ chelators such as EDTA and EGTA, inhibiting cysteine in the catalytic core region using cystamine (CTM), and mimicking the substrate glutamine using a 6-Diazo-5-Oxo-L-Norleucine (DON). TG2 is active in the extracellular matrix, plasma membrane, cytoplasm, mitochondria, recycling endosomes, and nucleus, and it crosslinks various substrate proteins [31]. TG2 plays an important role in apoptosis, cell survival, cytoskeletal function, cell motility, adhesion, and signal transduction. The opposing roles of TG2 have been reported in the brain, liver, cancer, pancreas, and angiogenesis [32].
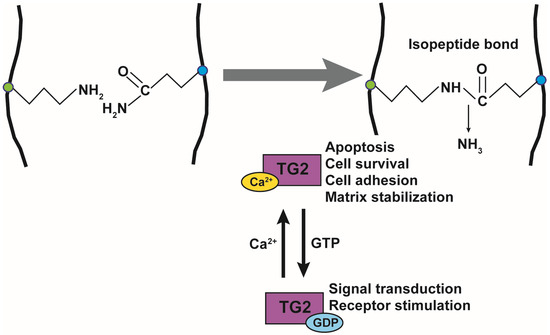
Figure 1. Transglutaminase 2 activity and function. Transamidation activity of transglutaminase 2 (TG2) is activated by Ca2+ and catalyzes the formation of an isopeptide bond. TG2 function is also regulated by guanosine triphosphate (GTP) binding. Ca2+-binding to TG2 regulates apoptosis, cell survival, cell adhesion, and matrix stabilization, while the binding of GDP to TG2 regulates signal transduction and receptor activation.
TG exerts antibacterial and antiviral effects through its substrates. Elafin is a member of the Trappin family and has been reported to suppress the growth of Pseudomonas aeruginosa, infection and replication of human immunodeficiency virus 1 (HIV-1), and infection with herpes simplex virus 2 (HSV-2) [33][34][35]. Trappin has a signal sequence for secretion (Pre), a TG substrate domain, and an inhibitory region containing a whey acidic protein (WAP)-motif that shows antibacterial and antiviral activities. Elafin is expressed in the skin, trachea, and vagina, and it is efficiently crosslinked to the extracellular matrix (ECM) through the TG substrate domain, which consists of a repeating sequence with lysine (K) and glutamine (Q)-rich “KGQDPV” sequence. TG controls the localization of Trappin by covalent crosslinking, releasing the WAP-motif from ECM, which, in turn, increases its antibacterial and antiviral activities (Figure 2) [36]. Women demonstrating resistance to HIV-1 infection have been shown to have a higher expression of Elafin with only the 6 kDa WAP motif in the reproductive organs [35]. Recombinant Elafin and Trappin-2 inhibit HIV-1 replication in T cells [36][37]. The Trappin family was formed through gene duplication, especially in porcine relatives, and underwent accelerated evolution [38][39]. They are thought to function as part of the innate immune response to various bacteria and viruses.
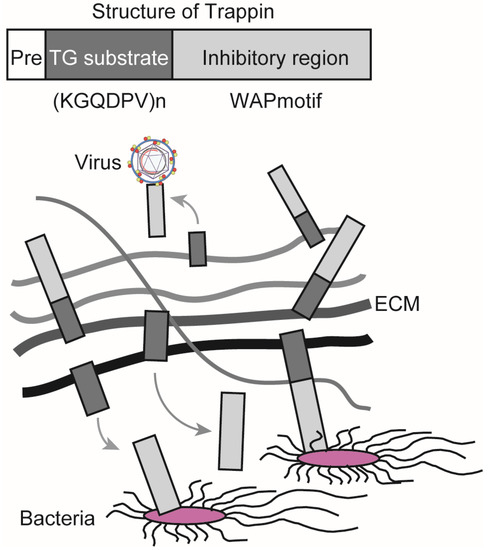
Figure 2. Trappin is responsible for antibacterial and antiviral activities. Trappin has a signal sequence for secretion (Pre), a TG substrate domain with a repeat sequence KGQDPV, and an inhibitory region that has a whey acidic protein (WAP) motif. Trappin is crosslinked to the extracellular matrix (ECM) by TG, whereby an inhibitory region is released into the extracellular space. The WAP motif exerts antibacterial and antiviral activities.
Our previous study showed that both the transaminase activity and expression of TG2 were increased in the livers of LPS-induced sepsis mice [22]. Single-cell RNA sequencing (scRNA-seq) provided a comprehensive view of TG2 gene expression in liver-composing cells [40]. Data mining of the scRNA-seq database and immunofluorescence staining revealed that the expression of TG2 in the liver under steady-state conditions was mainly detected in macrovascular and sinusoidal endothelial cells [22][40][41]. However, under inflammatory conditions, enhanced TG2 expression was observed in the F4/80-positive midzonal macrophages, suggesting that activated macrophages were the major cellular source of TG2 in the livers of sepsis mice [22]. The increase in TG2 gene expression in macrophages is believed to be involved in the activation cycle of the inflammatory process [19]. The stimulation of Toll-like receptor 4 (TLR4) by LPS induces the release of critical pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), which triggers a series of intracellular events that result in the activation of transcription factor nuclear factor-kappa B (NF-κB) and the activation of mitogen-activated protein kinases (MAPKs) [42][43]. The gene expression of TG2 is directly regulated by NF-κB through the NF-κB-binding motif in its promoter region [44][45]. In contrast, TG2 is also critical in NF-κB activation by promoting thrombin-induced DNA binding and serine phosphorylation of RelA/p65 [46] or by stimulating the polymerization of the inhibitory subunit α of NF-κB (I-κBα) [19][47]. The TG2-dependent activation of NF-κB might further promote the survival of macrophages [48] and a continuous activation cycle during the inflammatory process in sepsis.
The dysregulation of apoptosis in immune and nonimmune cells plays a critical role in the pathogenesis of sepsis [49][50]. Opposing roles of TG2 have been reported in the regulation of cell death and apoptosis [32]. TG2 gene expression in peripheral blood mononuclear cells and lymph nodes was induced following human immunodeficiency virus (HIV) infection [51]. An increased transaminase activity of TG2 was observed in lymphocytes undergoing apoptosis during the process of HIV infection [52]. Our group has shown that TG2 has a nuclear localization signal (NLS) and a nuclear export signal (NES). Its localization to the nucleus depends on importin, and its export from the nucleus depends on chromosomal region maintenance 1 (CRM1) [53]. TG2 was shown to translocate into the nucleus, and there was a crosslink that inactivated the transcription factor Sp1, inducing apoptosis by suppressing c-Met expression [54]. Following the co-culture of Candida albicans and hepatocytes, TG2 translocates into the nucleus and is activated by ROS generated by Candida albicans [55]. The induction of TG2 was also involved in the cell death induced by retinoids and fatty acids in hepatic cells [56][57][58]. In the LPS-induced sepsis mouse model, TG2 in the hepatic macrophages was activated, and the inhibition of TG2 activation prevented LPS-induced hepatic damage, as indicated by the serum level of ALT, which is a well-established indicator of hepatocyte cell death [22]. In contrast, the anti-apoptotic role of TG2 as a G protein has also been reported [59]. In TNF-α-induced sepsis mice, TG2 inhibited liver injury by downregulating TNF-α-induced expression of pro-death proteases caspase 3 and cathepsin D [60]. Thus, TG2 appears to be involved in host–pathogen interactions and plays multiple roles in the mechanisms of cell response to inflammatory substances in a context-dependent manner.
Celiac disease is a type of autoimmune disease that occurs primarily in the intestine, causing inflammatory disorders and villous atrophy triggered by gluten intake. Celiac disease affects approximately 1% of people in Europe and the United States [61]. It has been reported that antibodies against gliadin and TG2 are detected in patients with celiac disease [62][63]. In addition, Gln (Q) of gliadin is converted to Glu (E) by the deamination activity of TG2. This is thought to increase antigenicity, activate the immune system through the activation of T and B cells, and exacerbate autoimmune disease [64].
It has been reported that mucosal permeability increases in celiac disease because it causes inflammatory disorders in the small intestine [65], and that bacteria are more likely to bind to the small intestinal epithelium of celiac disease patients [66]. It has been suggested that the risk of causing sepsis is increased. Using Swedish national health resisters, Ludvigsson et al. showed that patients with celiac disease are more likely to develop sepsis, especially caused by pneumococcal and staphylococci, based on patient information that is not a celiac disease or celiac disease [67]. Thus, sepsis is also associated with celiac disease, which is exacerbated by the deamination of gliadin by TG2. This suggests that suppression by a TG2-specific inhibitor works effectively to treat celiac disease. However, a TG2-specific inhibitor could induce side effects because of the possible increased membrane permeability of the gut.
3. Targeting TG2 in Inflammation and Sepsis: Evidence from Knockout Mouse Models
Targeted mutant mice, such as those with a gene knockout (KO), play a vital role in understanding the integrated physiological and pathological functions of a protein in a tissue-specific context [68]. Three TG2 KO mouse lines [69][70][71] were generated using genomic DNA from the 129 sub-strain to target the catalytic domain of the Tgm2 gene. These genetically engineered mouse models have been used to explore the biological roles of TG2 in diverse pathophysiological contexts (reviewed in [72]). Under physiological conditions, TG2 KO mice were viable, with no obvious phenotypic or developmental abnormality compared to wild-type (WT) mice [69][70][71]. This could be partly explained by the compensatory activation, either transcriptional upregulation or upregulation of transamidation activity, of other TGs [73][74]. Under pathological conditions, a reduced inflammatory response has been reported in TG2 KO mice. In the pulmonary fibrosis [75] and allergic asthma model [76], reduced inflammation was observed in the TG2 KO mice compared to WT mice, which was possibly due to the regulation of T helper cells and the recruitment of both innate and adaptive immune cells. In a Parkinson’s disease (PD) model, the TG2 KO mice reversed the behavioral manifestations of PD by downregulating the release of inflammatory mediators such as histamine, leukotrienes, and cytokines by mast cells in the substantia nigra, suggesting that TG2 might contribute to neuroinflammation and neurodegeneration [77]. In a multiple sclerosis (MS) model, TG2 was present in the major histocompatibility complex class II-positive infiltrating cells in active MS lesions [78]. TG2 KO mice showed reduced experimental autoimmune encephalomyelitis, supporting a role of TG2 in MS pathogenesis [76][78]. The underlying mechanism was related with the inhibition of influx of immunomodulatory macrophages into the central nervous system [78]. In the UV irradiation-induced skin inflammation model, the TG2 KO mice showed reduced pro-inflammatory cytokine production in the keratinocytes [79]. Mechanistically, UV irradiation induced the activation of phospholipase C and calcium release from the endoplasmic reticulum, which leads to TG2 activation but does not induce TG2 gene expression [79]. On the other hand, an anti-inflammatory effect was also reported in TG2 KO mice. In a peritonitis model, TG2 KO mice showed hyperinflammation after exposure to monosodium urate (MSU) crystals [80]. In vitro mechanistic analysis revealed that TG2 overexpression suppressed MSU crystal-induced IL-1β and TNF-α production in macrophages through a transforming growth factor-β (TGF-β)-dependent pathway [80].
The role of TG2 deficiency in the pathology of sepsis is controversial. Following LPS-induced inflammation, a decrease in NF-κΒ activation and the sequestration of polymorphonuclear leukocytes (PMNs) was observed in the lungs of TG2 KO mice, suggesting that TG2 promotes endothelial cell inflammation and lung PMN infiltration [46]. In TNF-α-dependent septic mice, TG2 exhibited a protective role in liver injury, and TG2 KO mice showed increased liver damage compared to WT mice [60]. However, it has also been reported that TG2 KO mice improve survival after LPS-induced septic shock compared to WT mice. A marked reduction in the inflammatory response and attenuated organ damage, partly through the regulation of DC differentiation and function, was observed in the TG2 KO mice [19][21]. One explanation for these inconsistencies is that targeted mutations might have different phenotypes in different strain backgrounds, which, in combination with the difference in study design, significantly influences the reliability and reproducibility of the studies. Another explanation is that it is likely that the effect of TG2 on the progression of inflammation and sepsis may depend on the targets present in different cells and tissues, such as the apoptotic stress in epithelial cells and inflammatory stress in immune cells. Indeed, a dual role for TG2 has been reported to promote and protect the pathology of liver injury and obesity-related inflammation [41][54][81][82].
Given the critical roles of genetic knockout animal models in exploring the function of TG2 in the impairment of cell response in the presence of sepsis inducers, further studies using genetically engineered mouse models with tissue-specific deletion of Tgm2 are required to explore the cell type and disease stage-dependent roles of TG2 in inflammation and sepsis. As proof of principle, endothelial cell-specific deletion of TG2 provided direct evidence demonstrating TG2 controls allergic inflammation by regulating the recruitment of eosinophils into the lung endothelium [83].
4. Targeting TG2 in Inflammation and Sepsis: Evidence from Pharmacological Inhibition
Given the critical role of TG2 in a wide range of physiological disorders, the development of TG2 inhibitors has been the subject of intense research. CTM is a symmetric disulfide compound and is one of the earliest known pharmacological inhibitors of TG2, acting via competitive amine inhibition [84] and irreversible oxidation [85]. There is abundant evidence indicating that the suppression of TG2 transamidation with CTM has a beneficial effect against inflammation. CTM significantly delayed neuroinflammation in amyotrophic lateral sclerosis by inhibiting the TG2-induced oligomerization of superoxide dismutase 1 [86]. In a rat model of inflammatory bowel disease, CTM reduced the severity of colitis, which was associated with a decrease in TG2 activity and the production of pro-inflammatory cytokines such as TNF-α and IL-6 [87]. During lung inflammation in cystic fibrosis (CF), CTM restored TG2-mediated crosslinking of beclin 1 and subsequently rescued defective autophagy, aggresome formation, and the CF airway phenotype [88]. In contrast to its well-known anti-inflammatory effect, little is known about the effect of CTM on sepsis. Recently, we established an ex vivo imaging system to detect the in vivo transamidation activity of TG2 based on the incorporation of a biotinylated substrate for TG2, 5-biotinamidopentylamine (5BAPA), in the liver of LPS and cecal ligation and puncture (CLP)-induced sepsis mouse model (Figure 3A,B) [22]. We found that an LPS challenge and CLP operation dramatically induced the expression and activation of TG2 in the midzonal F4/80/CD80-positive M1 macrophages in the murine liver. The administration of CTM, 30 min before the LPS challenge, almost completely suppressed the 5BAPA signals in the liver and ameliorated the LPS-induced liver injury, suggesting that targeting TG2 with CTM holds great promise as a therapeutic strategy for sepsis (Figure 3C,D) [22]. Interestingly, pharmacological inhibition of TG2 with CTM at an early stage and not the late stage alleviated Schistosoma japonicum-induced liver fibrosis [89]. In addition to their therapeutic impact, pharmacological inhibitors such as CTM could be used as mechanistic probes to explore the disease stage-dependent physiological and pathological roles of TG2.
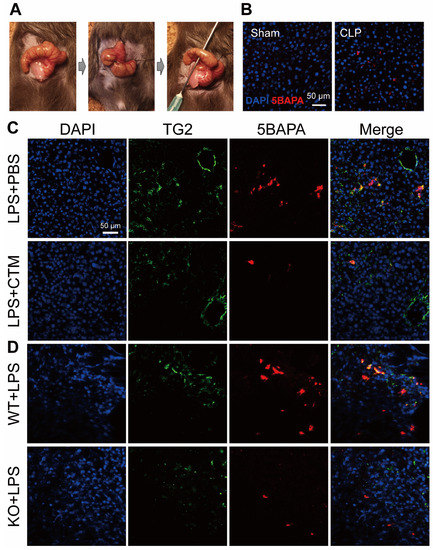
Figure 3. TG2 activity in the liver of septic mice. (A) Representative images from the cecal ligation and puncture (CLP) procedures. (B) TG2 activity in the liver of CLP-operated mice. Immunofluorescence staining with 4′,6-diamidino-2-phenylindole (DAPI) (blue) and 5-biotinamidopentylamine (5BAPA) (red) in frozen liver sections obtained from mice 48 h after sham or CLP operations. Scale bar: 50 μm. (C) TG2 activity in the liver of lipopolysaccharides (LPS)-injected mice. Immunofluorescence staining with DAPI (blue), TG2 (green), and 5BAPA (red) in frozen liver sections obtained from mice injected intraperitoneally with LPS in the presence and absence of CTM. Scale bar: 50 μm. (D) TG2 activity in the liver of LPS-injected TG2 knockout (KO) mice. Immunofluorescence staining of DAPI (blue), TG2 (green), and 5BAPA (red) in frozen liver sections obtained from mice injected intraperitoneally with 10 mg/kg LPS for 24 h. Scale bar: 50 μm. These images have been adapted from a previous study [22].
However, it should be noted that the inhibitory effect of CTM on transamidation activity is not specific to TG2 [90]. In addition, CTM can interact with other signaling pathways involved in the regulation of cell proliferation and death, such as the caspase signaling pathway [91]. Therefore, recent research has focused on the development of selective inhibitors against TG2 for their therapeutic potential (reviewed in [92]). These novel irreversible inhibitors of TG2, such as NC9 and VA4 [93], which specifically react with Cys-277 in the TG2 transamidation site and inhibit both transamidation and GTP-binding activities, are potent anticancer agents that inhibit the survival of cancer stem cells [94]. Very recently, an in vitro study showed that NC9 effectively reduced the levels of pro-inflammatory cytokines such as monocyte chemotactic protein 1, IL-1β, and TNF-α, in combined all-trans retinoic acid and arsenic trioxide-treated acute promyelocytic leukemia cells [95].
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). Jama 2016, 315, 801–810.
- Finfer, S.; Machado, F.R. The Global Epidemiology of Sepsis. Does It Matter That We Know So Little? Am. J. Respir. Crit. Care Med. 2016, 193, 228–230.
- Fleischmann, C.; Scherag, A.; Adhikari, N.K.; Hartog, C.S.; Tsaganos, T.; Schlattmann, P.; Angus, D.C.; Reinhart, K. Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current Estimates and Limitations. Am. J. Respir. Crit. Care Med. 2016, 193, 259–272.
- Reinhart, K.; Daniels, R.; Kissoon, N.; Machado, F.R.; Schachter, R.D.; Finfer, S. Recognizing Sepsis as a Global Health Priority—A WHO Resolution. N. Engl. J. Med. 2017, 377, 414–417.
- Murray, C.J.; Lopez, A.D. Measuring the global burden of disease. N. Engl. J. Med. 2013, 369, 448–457.
- Vincent, J.L.; Sakr, Y.; Sprung, C.L.; Ranieri, V.M.; Reinhart, K.; Gerlach, H.; Moreno, R.; Carlet, J.; Le Gall, J.R.; Payen, D. Sepsis in European intensive care units: Results of the SOAP study. Crit. Care Med. 2006, 34, 344–353.
- Zahar, J.R.; Timsit, J.F.; Garrouste-Orgeas, M.; Français, A.; Vesin, A.; Descorps-Declere, A.; Dubois, Y.; Souweine, B.; Haouache, H.; Goldgran-Toledano, D.; et al. Outcomes in severe sepsis and patients with septic shock: Pathogen species and infection sites are not associated with mortality. Crit. Care Med. 2011, 39, 1886–1895.
- Bhatraju, P.K.; Ghassemieh, B.J.; Nichols, M.; Kim, R.; Jerome, K.R.; Nalla, A.K.; Greninger, A.L.; Pipavath, S.; Wurfel, M.M.; Evans, L.; et al. Covid-19 in Critically Ill Patients in the Seattle Region—Case Series. N. Engl. J. Med. 2020, 382, 2012–2022.
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720.
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506.
- Delano, M.J.; Ward, P.A. Sepsis-induced immune dysfunction: Can immune therapies reduce mortality? J. Clin. Investig. 2016, 126, 23–31.
- Marshall, J.C. Why have clinical trials in sepsis failed? Trends Mol. Med. 2014, 20, 195–203.
- Mebazaa, A.; Laterre, P.F.; Russell, J.A.; Bergmann, A.; Gattinoni, L.; Gayat, E.; Harhay, M.O.; Hartmann, O.; Hein, F.; Kjolbye, A.L.; et al. Designing phase 3 sepsis trials: Application of learned experiences from critical care trials in acute heart failure. J. Intensive Care 2016, 4, 24.
- Peters van Ton, A.M.; Kox, M.; Abdo, W.F.; Pickkers, P. Precision Immunotherapy for Sepsis. Front. Immunol. 2018, 9, 1926.
- Cecconi, M.; Evans, L.; Levy, M.; Rhodes, A. Sepsis and septic shock. Lancet 2018, 392, 75–87.
- Odii, B.O.; Coussons, P. Biological functionalities of transglutaminase 2 and the possibility of its compensation by other members of the transglutaminase family. Sci. World J. 2014, 2014, 714561.
- Chrobok, N.L.; Sestito, C.; Wilhelmus, M.M.; Drukarch, B.; van Dam, A.M. Is monocyte- and macrophage-derived tissue transglutaminase involved in inflammatory processes? Amino Acids 2017, 49, 441–452.
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003, 4, 140–156.
- Falasca, L.; Farrace, M.G.; Rinaldi, A.; Tuosto, L.; Melino, G.; Piacentini, M. Transglutaminase type II is involved in the pathogenesis of endotoxic shock. J. Immunol. 2008, 180, 2616–2624.
- Jeong, E.M.; Son, Y.H.; Choi, Y.; Kim, J.H.; Lee, J.H.; Cho, S.Y.; Kim, I.G. Transglutaminase 2 is dispensable but required for the survival of mice in dextran sulfate sodium-induced colitis. Exp. Mol. Med. 2016, 48, e267.
- Matic, I.; Sacchi, A.; Rinaldi, A.; Melino, G.; Khosla, C.; Falasca, L.; Piacentini, M. Characterization of transglutaminase type II role in dendritic cell differentiation and function. J. Leukoc. Biol. 2010, 88, 181–188.
- Su, T.; Qin, X.Y.; Furutani, Y.; Yu, W.; Kojima, S. Imaging of the ex vivo transglutaminase activity in liver macrophages of sepsis mice. Anal. Biochem. 2020, 597, 113654.
- Szondy, Z.; Korponay-Szabó, I.; Király, R.; Sarang, Z.; Tsay, G.J. Transglutaminase 2 in human diseases. BioMedicine 2017, 7, 15.
- Nemes, Z.; Steinert, P.M. Bricks and mortar of the epidermal barrier. Exp. Mol. Med. 1999, 31, 5–19.
- Matsuki, M.; Yamashita, F.; Ishida-Yamamoto, A.; Yamada, K.; Kinoshita, C.; Fushiki, S.; Ueda, E.; Morishima, Y.; Tabata, K.; Yasuno, H.; et al. Defective stratum corneum and early neonatal death in mice lacking the gene for transglutaminase 1 (keratinocyte transglutaminase). Proc. Natl. Acad. Sci. USA 1998, 95, 1044–1049.
- Kuramoto, N.; Takizawa, T.; Takizawa, T.; Matsuki, M.; Morioka, H.; Robinson, J.M.; Yamanishi, K. Development of ichthyosiform skin compensates for defective permeability barrier function in mice lacking transglutaminase 1. J. Clin. Investig. 2002, 109, 243–250.
- Piro, M.C.; Ventura, A.; Smirnov, A.; Saggini, A.; Lena, A.M.; Mauriello, A.; Bianchi, L.; Melino, G.; Candi, E. Transglutaminase 3 Reduces the Severity of Psoriasis in Imiquimod-Treated Mouse Skin. Int. J. Mol. Sci. 2020, 21, 1566.
- Candi, E.; Oddi, S.; Paradisi, A.; Terrinoni, A.; Ranalli, M.; Teofoli, P.; Citro, G.; Scarpato, S.; Puddu, P.; Melino, G. Expression of transglutaminase 5 in normal and pathologic human epidermis. J. Investig. Dermatol. 2002, 119, 670–677.
- Furutani, Y.; Kato, A.; Fibriani, A.; Hirata, T.; Kawai, R.; Jeon, J.H.; Fujii, Y.; Kim, I.G.; Kojima, S.; Hirose, S. Identification, evolution, and regulation of expression of Guinea pig trappin with an unusually long transglutaminase substrate domain. J. Biol. Chem. 2005, 280, 20204–20215.
- Mehta, K.; Eckert, R. Transglutaminases: Family of Enzymes with Diverse Functions; Karger: Basel, Switzerland, 2005; Volume 38, p. 253.
- Hitomi, K.; Kojima, S.; Fesus, L. Transglutaminases, Multiple Functional Modifiers and Targets for New Drug Discovery, 1st ed.; Springer: Tokyo, Japan, 2015; p. VIII, 391.
- Tatsukawa, H.; Furutani, Y.; Hitomi, K.; Kojima, S. Transglutaminase 2 has opposing roles in the regulation of cellular functions as well as cell growth and death. Cell Death Dis. 2016, 7, e2244.
- Bellemare, A.; Vernoux, N.; Morisset, D.; Bourbonnais, Y. Human pre-elafin inhibits a Pseudomonas aeruginosa-secreted peptidase and prevents its proliferation in complex media. Antimicrob. Agents Chemother. 2008, 52, 483–490.
- Drannik, A.G.; Nag, K.; Sallenave, J.M.; Rosenthal, K.L. Antiviral activity of trappin-2 and elafin in vitro and in vivo against genital herpes. J. Virol. 2013, 87, 7526–7538.
- Iqbal, S.M.; Ball, T.B.; Levinson, P.; Maranan, L.; Jaoko, W.; Wachihi, C.; Pak, B.J.; Podust, V.N.; Broliden, K.; Hirbod, T.; et al. Elevated elafin/trappin-2 in the female genital tract is associated with protection against HIV acquisition. AIDS 2009, 23, 1669–1677.
- Drannik, A.G.; Nag, K.; Yao, X.D.; Henrick, B.M.; Jain, S.; Ball, T.B.; Plummer, F.A.; Wachihi, C.; Kimani, J.; Rosenthal, K.L. Anti-HIV-1 activity of elafin is more potent than its precursor’s, trappin-2, in genital epithelial cells. J. Virol. 2012, 86, 4599–4610.
- Jasinghe, V.J.; Peyrotte, E.A.; Meyers, A.F.; Gajanayaka, N.; Ball, T.B.; Sandstrom, P.; Lavigne, C. Human rElafin Inhibits HIV-1 Replication in Its Natural Target Cells. Biores. Open Access 2013, 2, 128–137.
- Furutani, Y.; Kato, A.; Yasue, H.; Alexander, L.J.; Beattie, C.W.; Hirose, S. Evolution of the trappin multigene family in the Suidae. J. Biochem. 1998, 124, 491–502.
- Kato, A.; Rooney, A.P.; Furutani, Y.; Hirose, S. Evolution of trappin genes in mammals. BMC Evolut. Biol. 2010, 10, 31.
- Aizarani, N.; Saviano, A.; Sagar; Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grün, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204.
- Piacentini, M.; Baiocchini, A.; Del Nonno, F.; Melino, G.; Barlev, N.A.; Rossin, F.; D’Eletto, M.; Falasca, L. Non-alcoholic fatty liver disease severity is modulated by transglutaminase type 2. Cell Death Dis. 2018, 9, 257.
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635.
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151.
- Kuncio, G.S.; Tsyganskaya, M.; Zhu, J.; Liu, S.L.; Nagy, L.; Thomazy, V.; Davies, P.J.; Zern, M.A. TNF-alpha modulates expression of the tissue transglutaminase gene in liver cells. Am. J. Physiol. 1998, 274, G240–G245.
- Mirza, A.; Liu, S.L.; Frizell, E.; Zhu, J.; Maddukuri, S.; Martinez, J.; Davies, P.; Schwarting, R.; Norton, P.; Zern, M.A. A role for tissue transglutaminase in hepatic injury and fibrogenesis, and its regulation by NF-kappaB. Am. J. Physiol. 1997, 272, G281–G288.
- Bijli, K.M.; Kanter, B.G.; Minhajuddin, M.; Leonard, A.; Xu, L.; Fazal, F.; Rahman, A. Regulation of endothelial cell inflammation and lung polymorphonuclear lymphocyte infiltration by transglutaminase 2. Shock 2014, 42, 562–569.
- Lee, J.; Kim, Y.S.; Choi, D.H.; Bang, M.S.; Han, T.R.; Joh, T.H.; Kim, S.Y. Transglutaminase 2 induces nuclear factor-kappaB activation via a novel pathway in BV-2 microglia. J. Biol. Chem. 2004, 279, 53725–53735.
- Lombardo, E.; Alvarez-Barrientos, A.; Maroto, B.; Boscá, L.; Knaus, U.G. TLR4-mediated survival of macrophages is MyD88 dependent and requires TNF-alpha autocrine signalling. J. Immunol. 2007, 178, 3731–3739.
- Hotchkiss, R.S.; Tinsley, K.W.; Swanson, P.E.; Schmieg, R.E., Jr.; Hui, J.J.; Chang, K.C.; Osborne, D.F.; Freeman, B.D.; Cobb, J.P.; Buchman, T.G.; et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J. Immunol. 2001, 166, 6952–6963.
- Mahidhara, R.; Billiar, T.R. Apoptosis in sepsis. Crit. Care Med. 2000, 28 (Suppl. S4), N105–N113.
- Amendola, A.; Rodolfo, C.; Di Caro, A.; Ciccosanti, F.; Falasca, L.; Piacentini, M. “Tissue” transglutaminase expression in HIV-infected cells: An enzyme with an antiviral effect? Ann. N. Y. Acad. Sci. 2001, 946, 108–120.
- Amendola, A.; Gougeon, M.L.; Poccia, F.; Bondurand, A.; Fesus, L.; Piacentini, M. Induction of “tissue” transglutaminase in HIV pathogenesis: Evidence for high rate of apoptosis of CD4+ T lymphocytes and accessory cells in lymphoid tissues. Proc. Natl. Acad. Sci. USA 1996, 93, 11057–11062.
- Shrestha, R.; Tatsukawa, H.; Shrestha, R.; Ishibashi, N.; Matsuura, T.; Kagechika, H.; Kose, S.; Hitomi, K.; Imamoto, N.; Kojima, S. Molecular mechanism by which acyclic retinoid induces nuclear localization of transglutaminase 2 in human hepatocellular carcinoma cells. Cell Death Dis. 2015, 6, e2002.
- Tatsukawa, H.; Fukaya, Y.; Frampton, G.; Martinez-Fuentes, A.; Suzuki, K.; Kuo, T.F.; Nagatsuma, K.; Shimokado, K.; Okuno, M.; Wu, J.; et al. Role of transglutaminase 2 in liver injury via cross-linking and silencing of transcription factor Sp1. Gastroenterology 2009, 136, 1783–1795.e10.
- Shrestha, R.; Shrestha, R.; Qin, X.Y.; Kuo, T.F.; Oshima, Y.; Iwatani, S.; Teraoka, R.; Fujii, K.; Hara, M.; Li, M.; et al. Fungus-derived hydroxyl radicals kill hepatic cells by enhancing nuclear transglutaminase. Sci. Rep. 2017, 7, 4746.
- Qin, X.Y.; Fujii, S.; Shimizu, A.; Kagechika, H.; Kojima, S. Carboxylic Derivatives of Vitamin K2 Inhibit Hepatocellular Carcinoma Cell Growth through Caspase/Transglutaminase-Related Signaling Pathways. J. Nutr. Sci. Vitaminol. 2015, 61, 285–290.
- Qin, X.Y.; Lu, J.; Cai, M.; Kojima, S. Arachidonic acid suppresses hepatic cell growth through ROS-mediated activation of transglutaminase. FEBS Open Bio 2018, 8, 1703–1710.
- Qin, X.Y.; Su, T.; Kojima, S. Prevention of arachidonic acid-induced liver injury by controlling oxidative stress-mediated transglutaminase activation with garlic extracts. Exp. Ther. Med. 2020, 19, 1522–1527.
- Sarang, Z.; Molnár, P.; Németh, T.; Gomba, S.; Kardon, T.; Melino, G.; Cotecchia, S.; Fésüs, L.; Szondy, Z. Tissue transglutaminase (TG2) acting as G protein protects hepatocytes against Fas-mediated cell death in mice. Hepatology 2005, 42, 578–587.
- Yoo, H.; Ahn, E.R.; Kim, S.J.; Lee, S.H.; Oh, S.H.; Kim, S.Y. Divergent results induced by different types of septic shock in transglutaminase 2 knockout mice. Amino Acids 2013, 44, 189–197.
- Lebwohl, B.; Sanders, D.S.; Green, P.H.R. Coeliac disease. Lancet 2018, 391, 70–81.
- Aleanzi, M.; Demonte, A.M.; Esper, C.; Garcilazo, S.; Waggener, M. Celiac disease: Antibody recognition against native and selectively deamidated gliadin peptides. Clin. Chem. 2001, 47, 2023–2028.
- Dieterich, W.; Laag, E.; Schöpper, H.; Volta, U.; Ferguson, A.; Gillett, H.; Riecken, E.O.; Schuppan, D. Autoantibodies to tissue transglutaminase as predictors of celiac disease. Gastroenterology 1998, 115, 1317–1321.
- Fleckenstein, B.; Molberg, Ø.; Qiao, S.W.; Schmid, D.G.; von der Mülbe, F.; Elgstøen, K.; Jung, G.; Sollid, L.M. Gliadin T cell epitope selection by tissue transglutaminase in celiac disease. Role of enzyme specificity and pH influence on the transamidation versus deamidation process. J. Biol. Chem. 2002, 277, 34109–34116.
- Doig, C.J.; Sutherland, L.R.; Sandham, J.D.; Fick, G.H.; Verhoef, M.; Meddings, J.B. Increased intestinal permeability is associated with the development of multiple organ dysfunction syndrome in critically ill ICU patients. Am. J. Respir. Crit. Care Med. 1998, 158, 444–451.
- Forsberg, G.; Fahlgren, A.; Hörstedt, P.; Hammarström, S.; Hernell, O.; Hammarström, M.L. Presence of bacteria and innate immunity of intestinal epithelium in childhood celiac disease. Am. J. Gastroenterol. 2004, 99, 894–904.
- Ludvigsson, J.F.; Olén, O.; Bell, M.; Ekbom, A.; Montgomery, S.M. Coeliac disease and risk of sepsis. Gut 2008, 57, 1074–1080.
- Iismaa, S.E. Insights into Transglutaminase 2 Function Gained from Genetically Modified Animal Models; Transglutaminases; Hitomi, K., Kojima, S., Fesus, L., Eds.; Springer: Tokyo, Japan, 2015; pp. 83–115.
- De Laurenzi, V.; Melino, G. Gene disruption of tissue transglutaminase. Mol. Cell Biol. 2001, 21, 148–155.
- Kim, D.S.; Kim, B.; Tahk, H.; Kim, D.H.; Ahn, E.R.; Choi, C.; Jeon, Y.; Park, S.Y.; Lee, H.; Oh, S.H.; et al. Transglutaminase 2 gene ablation protects against renal ischemic injury by blocking constant NF-κB activation. Biochem. Biophys. Res. Commun. 2010, 403, 479–484.
- Nanda, N.; Iismaa, S.E.; Owens, W.A.; Husain, A.; Mackay, F.; Graham, R.M. Targeted inactivation of Gh/tissue transglutaminase II. J. Biol. Chem. 2001, 276, 20673–20678.
- Iismaa, S.E.; Mearns, B.M.; Lorand, L.; Graham, R.M. Transglutaminases and disease: Lessons from genetically engineered mouse models and inherited disorders. Physiol. Rev. 2009, 89, 991–1023.
- Deasey, S.; Shanmugasundaram, S.; Nurminskaya, M. Tissue-specific responses to loss of transglutaminase 2. Amino Acids 2013, 44, 179–187.
- Johnson, K.B.; Petersen-Jones, H.; Thompson, J.M.; Hitomi, K.; Itoh, M.; Bakker, E.N.; Johnson, G.V.; Colak, G.; Watts, S.W. Vena cava and aortic smooth muscle cells express transglutaminases 1 and 4 in addition to transglutaminase 2. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1355–H1366.
- Oh, K.; Park, H.B.; Byoun, O.J.; Shin, D.M.; Jeong, E.M.; Kim, Y.W.; Kim, Y.S.; Melino, G.; Kim, I.G.; Lee, D.S. Epithelial transglutaminase 2 is needed for T cell interleukin-17 production and subsequent pulmonary inflammation and fibrosis in bleomycin-treated mice. J. Exp. Med. 2011, 208, 1707–1719.
- Oh, K.; Park, H.B.; Seo, M.W.; Byoun, O.J.; Lee, D.S. Transglutaminase 2 exacerbates experimental autoimmune encephalomyelitis through positive regulation of encephalitogenic T cell differentiation and inflammation. Clin. Immunol. 2012, 145, 122–132.
- Hong, G.U.; Cho, J.W.; Kim, S.Y.; Shin, J.H.; Ro, J.Y. Inflammatory mediators resulting from transglutaminase 2 expressed in mast cells contribute to the development of Parkinson’s disease in a mouse model. Toxicol. Appl. Pharmacol. 2018, 358, 10–22.
- Van Strien, M.E.; de Vries, H.E.; Chrobok, N.L.; Bol, J.; Breve, J.J.P.; van der Pol, S.M.P.; Kooij, G.; van Buul, J.D.; Karpuj, M.; Steinman, L.; et al. Tissue Transglutaminase contributes to experimental multiple sclerosis pathogenesis and clinical outcome by promoting macrophage migration. Brain Behav. Immun. 2015, 50, 141–154.
- Lee, S.J.; Lee, K.B.; Son, Y.H.; Shin, J.; Lee, J.H.; Kim, H.J.; Hong, A.Y.; Bae, H.W.; Kwon, M.A.; Lee, W.J.; et al. Transglutaminase 2 mediates UV-induced skin inflammation by enhancing inflammatory cytokine production. Cell Death Dis. 2017, 8, e3148.
- Yen, J.H.; Lin, L.C.; Chen, M.C.; Sarang, Z.; Leong, P.Y.; Chang, I.C.; Hsu, J.D.; Chen, J.H.; Hsieh, Y.F.; Pallai, A.; et al. The metastatic tumor antigen 1-transglutaminase-2 pathway is involved in self-limitation of monosodium urate crystal-induced inflammation by upregulating TGF-β1. Arthritis Res. Ther. 2015, 17, 65.
- Cassiman, D.; Libbrecht, L.; Desmet, V.; Denef, C.; Roskams, T. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J. Hepatol. 2002, 36, 200–209.
- Sághy, T.; Köröskényi, K.; Hegedűs, K.; Antal, M.; Bankó, C.; Bacsó, Z.; Papp, A.; Stienstra, R.; Szondy, Z. Loss of transglutaminase 2 sensitizes for diet-induced obesity-related inflammation and insulin resistance due to enhanced macrophage c-Src signaling. Cell Death Dis. 2019, 10, 439.
- Soveg, F.; Abdala-Valencia, H.; Campbell, J.; Morales-Nebreda, L.; Mutlu, G.M.; Cook-Mills, J.M. Regulation of allergic lung inflammation by endothelial cell transglutaminase 2. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L573–L583.
- Jeitner, T.M.; Delikatny, E.J.; Ahlqvist, J.; Capper, H.; Cooper, A.J. Mechanism for the inhibition of transglutaminase 2 by cystamine. Biochem. Pharmacol. 2005, 69, 961–970.
- Palanski, B.A.; Khosla, C. Cystamine and Disulfiram Inhibit Human Transglutaminase 2 via an Oxidative Mechanism. Biochemistry 2018, 57, 3359–3363.
- Oono, M.; Okado-Matsumoto, A.; Shodai, A.; Ido, A.; Ohta, Y.; Abe, K.; Ayaki, T.; Ito, H.; Takahashi, R.; Taniguchi, N.; et al. Transglutaminase 2 accelerates neuroinflammation in amyotrophic lateral sclerosis through interaction with misfolded superoxide dismutase 1. J. Neurochem. 2014, 128, 403–418.
- Elli, L.; Ciulla, M.M.; Busca, G.; Roncoroni, L.; Maioli, C.; Ferrero, S.; Bardella, M.T.; Bonura, A.; Paliotti, R.; Terrani, C.; et al. Beneficial effects of treatment with transglutaminase inhibitor cystamine on the severity of inflammation in a rat model of inflammatory bowel disease. Lab. Investig. J. Tech. Methods Pathol. 2011, 91, 452–461.
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12, 863–875.
- Wen, Z.; Ji, X.; Tang, J.; Lin, G.; Xiao, L.; Liang, C.; Wang, M.; Su, F.; Ferrandon, D.; Li, Z. Positive Feedback Regulation between Transglutaminase 2 and Toll-Like Receptor 4 Signaling in Hepatic Stellate Cells Correlates with Liver Fibrosis Post Schistosoma japonicum Infection. Front. Immunol. 2017, 8, 1808.
- Siegel, M.; Khosla, C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol. Ther. 2007, 115, 232–245.
- Lesort, M.; Lee, M.; Tucholski, J.; Johnson, G.V. Cystamine inhibits caspase activity. Implications for the treatment of polyglutamine disorders. J. Biol. Chem. 2003, 278, 3825–3830.
- Keillor, J.W.; Apperley, K.Y.; Akbar, A. Inhibitors of tissue transglutaminase. Trends Pharmacol. Sci. 2015, 36, 32–40.
- Akbar, A.; McNeil, N.M.R.; Albert, M.R.; Ta, V.; Adhikary, G.; Bourgeois, K.; Eckert, R.L.; Keillor, J.W. Structure-Activity Relationships of Potent, Targeted Covalent Inhibitors That Abolish Both the Transamidation and GTP Binding Activities of Human Tissue Transglutaminase. J. Med. Chem. 2017, 60, 7910–7927.
- Kerr, C.; Szmacinski, H.; Fisher, M.L.; Nance, B.; Lakowicz, J.R.; Akbar, A.; Keillor, J.W.; Lok Wong, T.; Godoy-Ruiz, R.; Toth, E.A.; et al. Transamidase site-targeted agents alter the conformation of the transglutaminase cancer stem cell survival protein to reduce GTP binding activity and cancer stem cell survival. Oncogene 2017, 36, 2981–2990.
- Jambrovics, K.; Uray, I.P.; Keillor, J.W.; Fésüs, L.; Balajthy, Z. Benefits of Combined All-Trans Retinoic Acid and Arsenic Trioxide Treatment of Acute Promyelocytic Leukemia Cells and Further Enhancement by Inhibition of Atypically Expressed Transglutaminase 2. Cancers 2020, 12, 648.

