 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Irina Bogolyubova | + 2656 word(s) | 2656 | 2021-02-24 07:17:27 |
Video Upload Options
The Death-domain associated protein 6 (DAXX) is an evolutionarily conserved and ubiquitously expressed protein that is implicated in many cellular processes, including transcription, cellular proliferation, cell cycle regulation, Fas-induced apoptosis, and many other events.
1. Introduction
DAXX is a multifunctional protein present in the nucleus and cytoplasm (the cytoplasmic functions of DAXX are not discussed here). Mutations in the DAXX gene may be associated with some cancers [1], suggesting DAXX as one of the most important factors in maintaining the integrity of the genome. The nuclear function of DAXX is largely determined by its properties as a chaperone of the histone variant H3.3 [2][3]. DAXX-dependent deposition of H3.3 and known relations of the DAXX protein with transcription factors, epigenetic modifiers, and chromatin-remodeling proteins including the α-thalassemia/mental retardation syndrome X-linked protein ATRX [4] make DAXX a main player in chromatin silencing, mainly in the pericentromeric areas [5]. In this context, there is growing recent evidence that DAXX is an essential factor for proper development of mammalian oocytes and early embryos [6], since the processes of oocyte development during meiosis and fertilization, as well as the reprogramming of parental genomes in early embryos, are accompanied by pronounced rearrangements of the heterochromatin compartment [7][8], in which deposition of H3 variants plays an important role [9].
2. Structure and Localization
DAXX was identified in 1997 as a murine signaling protein, the C-terminal part of which specifically binds to the Fas death domain and enhances Fas-mediated apoptosis [10] by activating the Jun NH2-terminal kinase (JNK) pathway through the apoptosis signal-regulating kinase 1 (ASK1) [11]. In parallel, it has been shown that human DAXX (hDAXX), although specifically affecting Fas-mediated apoptosis, does not bind Fas and instead is found in the nucleus [12] due to the presence of two nucleus localization signals (NLS) in the molecule [13].
The structure of the DAXX molecule is now well established. A modular structure of the DAXX molecule and its main domains are depicted in Figure 1a. The molecule of DAXX exhibits six regions of sequence conservation, including a central histone-binding domain (HBD) [2], an N-terminal 4-helix bundle, and a C-terminal domain that is mostly disordered [14]. A combination of structural, biochemical, and cell-based targeting analyses of the H3.3/H4/DAXX HBD complex allowed revealing an extended fold of the DAXX HBD that envelops an H3.3/H4 dimer with seven consecutive α-helices [15][16]. The N- and C- termini of the DAXX molecule contain two SUMO-interacting motifs (SIMs: SIM-N and SIM-C, respectively), which both independently interact with a small ubiquitin-related modifier SUMO [17].

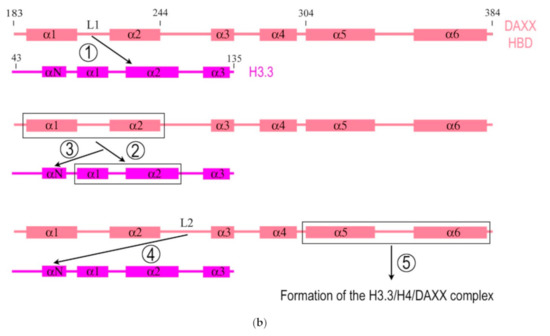
Figure 1. Simplified diagrams depicting the modular structure of the DAXX molecule (a) and interactions between the histone binding domain of DAXX and H3.3 during the formation of the complex H3.3/H4/DAXX (b). (a) DAXX contains two independent SUMO-interacting motifs at the N- and C-termini (SIM-N and SIM-C, respectively) [17]. The DAXX helix bundle serves as the main site for binding the α-thalassemia/mental retardation syndrome X-linked protein ATRX [18] and many other proteins that interact with the DAXX terminal region, including BRG1; another BRG1-interacting region is in the central part of DAXX [19]. The histone binding domain (HBD) is the main module involved in interactions with H3.3, a functional partner of DAXX, and the residue E225 in DAXX contributes to the specificity of this interaction. The HBD consists of six α-helices (α1–α6) and intervening loops (L1–L5); N-terminal helices (α1 and α2) are also termed ‘tower’ [15]. Two nucleus localization signals (NLS1 and NLS2), a Glu/Asp-rich acidic region, two segments rich, respectively, in Ser/Pro/Glu residues (SPE) and in Ser/Pro/Thr residues (SPT) are depicted. The most intrinsically disordered regions (IDRs) of the DAXX molecule are also shown. The C-terminus includes the regions to interact with the PML protein [20] and CENP-C [21], which impacts the peculiarities of DAXX distribution within the nucleus. The vast majority of DAXX interactions, not mentioned here, including those involved in cancer, are not depicted in the scheme, see [1] for additional reading. (b) DAXX is a specific H3.3 chaperone. The recognition and binding of H3.3 by the HBD of DAXX involve several folding steps [22], shown in the diagram by encircled numbers (1–5). 1—L1 of the DAXX ‘tower’ interacts with lateral H3.3 surface by long-range electrostatic interactions; 2–the ‘tower’ helices (α1 and α2) of DAXX, including portions of L1, fold onto two helical segments of H3.3 (α1 and α2), including the intervening loop (the interacting domains are ensquared); 3—the αN helix of H3.3 folds; 4—the L2 of DAXX tightly wraps around the folded H3.3 αN helix; 5—finally, the α5 and α6 helices of DAXX pack against H3.3 and H4, respectively, forming the H3.3/H4/DAXX complex. H4 interactions are not shown. For further reading and structural details regarding formation of the H3.3/H4/DAXX heterotrimer, see [15][22].
In the nucleus, SUMO-1, which modifies the promyelocytic leukemia protein (PML), is necessary for the recruitment of DAXX into the PML nuclear bodies (PML-NBs) [20], which play a role in regulation of transcription and support stability of the genome by sequestration, modification, and/or degradation of nuclear proteins [23]. The repressive function of DAXX to basal transcription is regulated by its interactions with the PML protein and is inhibited by overexpression of PML [24].
DAXX interacts with the centromere protein C (CENP-C), and this interaction is mediated by the N-terminal 315 amino acids of CENP-C and the C-terminal 104 amino acids of hDAXX [21]. At the same time, hDAXX was not found predominant in fractions obtained from partially purified mitotic chromosomes, indicating that hDAXX is either not a chromosomal protein during mitosis or that it readily dissociates from condensed mitotic chromosomes during biochemical treatments.
Importantly, DAXX interacts with the chromatin-remodeling protein ATRX—an ATP-dependent helicase, mutations in the gene of which cause the X-linked mental retardation syndrome associated with α-thalassemia [4]. ATRX and DAXX are both the components of the same ATP-dependent chromatin remodeling complex and localize to the PML-NBs. The level of these DAXX/ATRX complexes is reduced in cells of patients with ATRX syndrome [25].
DAXX interacts with the transcription activator BRG1—another chromatin-remodeling protein with an ATPase activity—and can serve as a negative regulator of several BRG1-regulated genes: either directly through BRG1 binding and/or indirectly via other factors [19]. It is possible that BRG1 and ATRX play a role in targeting of DAXX to specific chromatin regions, where DAXX performs its chromatin- and transcription-regulating functions.
3. DAXX Is an H3.3 Chaperone
Mammalian H3.3 is a variant of the major histone H3 (H3.1) that differs by only five amino acids [15]. H3.3 is deposited both at sites of active transcription with the participation of the histone cell cycle regulator A (HIRA) [26][27] and in the heterochromatic regions, including telomeres and pericentromeric zones, with participation of the DAXX/ATRX complex [2][28][29][30][31]. As an H3.3-specific chaperone, DAXX facilitates H3.3 deposition at H3K9me3-containing heterochromatin regions.
DAXX interacts directly with the H3.3/H4 heterodimer through its highly conserved HBD, which makes extensive contacts with both H3.3 and H4. The residues E225 in DAXX and G90 in H3.3 are the main determinants of chaperone-mediated H3.3 recognition specificity [15][16]. DAXX also uses a shallow hydrophobic pocket to accommodate the small hydrophobic A87 of H3.3, whereas a polar binding environment in DAXX prefers G90 in H3.3 over the hydrophobic M90 in H3.1 [16]. DAXX can specifically associate with H3.3/H4 despite a high concentration of nearly identical canonical H3 in the cell. According to the domain-level cooperative folding model (Figure 1b), a mostly unfolded DAXX initially makes contacts near the H3.3 specificity region and then can sample a large part of the H3.3 surface before folding into place. The dynamic stability of partially folded intermediates may be responsible for the discrimination of H3.3 from other H3 variants [22].
DAXX- and ATRX-mediated H3.3 chromatin assembly is required for many functions including H3K9 tri-methylation at pericentromeres, endogenous retroviral (ERV) sequences, imprinted genes, intragenic methylated CpG islands and telomeres in embryonic stem cells (ESCs) [32][33][34][35]. The function of DAXX as a H3.3 chaperone is mediated by its interaction with the constitutive centromeric protein CENP-B, which serves as a kind of ‘beacon’ for H3.3 incorporation [36]. The interaction of DAXX with CENP-B and the association of DAXX with centromeres are SUMO-dependent and require two SIMs of the DAXX molecule, enabling DAXX binding to SUMOylated proteins, such as PML [36]. Depletion of SUMO-2, but not SUMO-1, decreases the interaction between DAXX and CENP-B and impairs the accumulation of DAXX and H3.3 at centromeres, which proves the different functions of the SUMO paralogs in the H3.3 deposition processes [36]. DAXX-mediated H3.3 deposition is repressed by the PML protein, and PML-NBs coordinate this process [37]. Specifically, PML plays a role in the routing of H3.3 to chromatin and in the organization of megabase-size heterochromatic PML-associated domains (PADs). Loss of PML impairs the heterochromatic state of PADs by shifting the balance of H3 methylation from K9me3 to K27me3. In addition, this alters the ATRX/DAXX-dependent, but not HIRA-dependent, deposition of H3.3 in PADs [38].
DAXX not only regulates the deposition of H3.3, but is one of the leading factors in maintaining the global heterochromatin landscape, since loss of DAXX can seriously affect the subnuclear organization in general. In the absence of DAXX, there are changes in H3K9me3-enriched heterochromatin domains, including the loss of a typical chromocenter structure and the loss of overlap between non-nucleolar and perinucleolar compact chromatin and H3K9me3. In addition, the structural integrity of nucleoli and the organization of rDNA are disrupted [39].
DAXX is important for the regulation of the cell cycle, mitosis, and cytokinesis. A decrease in the S-phase duration accompanied by an increase in the number of cells with double nuclei, micronuclei, and nuclear blebs was observed in DAXX−/− cells [36][40], indicating mitotic segregation defects. DAXX can be involved in the control of mitosis in particular through the regulation of Aurora-A kinase and cyclin B stability, including through the interaction of DAXX with the mitotic checkpoint protein RASSF1 [41][42]. DAXX plays an important role in the regulation of centromeric heterochromatin activity. Knockout of DAXX reduces the association of ATRX with centromeres and significantly increases chromosomal instability [36][40]. Accumulation of DAXX in centromeric and pericentromeric chromatin regions is enhanced by stress, including heat shock, as shown for several cancer cell lines [43], suggesting that DAXX-containing complexes are useful in maintaining the normal epigenetic landscape of heterochromatin.
4. DAXX is Essential for Normal Development of Mammalian Oocytes and Embryos
DAXX demonstrates a significant role in global transformations of the nucleus during late oogenesis and early embryogenesis of mammals, but mostly, its function, which could impact on development, is still elusive. However, loss of DAXX in oocytes and embryos affects many nuclear processes (Figure 2).
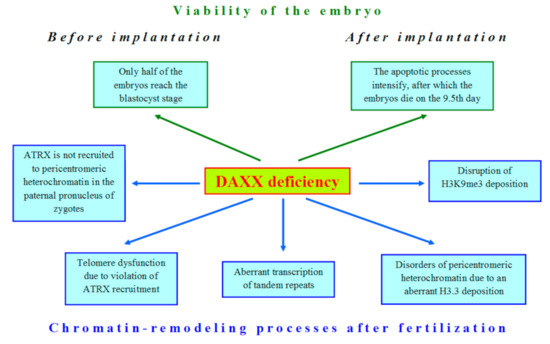
Figure 2. The deficiency of DAXX violates many cellular processes in early development of mammals.
The first attempts to shed light on the role of DAXX in embryonic development began immediately after the identification of this protein, when DAXX-deficient mice were obtained [44]. The authors expected to find a hyperproliferative disorder in development of these mice, but the result was unexpectedly opposite: mutations in the DAXX gene led to extensive apoptosis. Only 50% of DAXX-deficient embryos can reach the blastocyst stage [45]. After implantation, DAXX−/− embryos are developmentally retarded and completely disintegrated by the 12.5 day of gestation [40][44]. Since DAXX is an H3.3 chaperone, the disruption of the developmental program in DAXX-deficient embryos is not surprising due to the significant role of H3.3 in oogenesis and embryogenesis [46][47]. The whole-transcriptome analysis of metaphase II (MII) oocytes allowed revealing expression of DAXX and other H3.3 chaperones, such as HIRA, in mouse oogenesis [48]. However, DAXX cannot compensate normal H3.3 deposition in HIRA-depleted oocytes [49]. Similar results were also obtained for mouse ESCs [30].
In oocytes, DAXX predominantly localizes to the pericentromeric heterochromatin (PCH) regions, as shown by analysis of the nuclear localization of DAXX and centromeric proteins [50]. DAXX deposition in PCH is significantly reduced in fully-grown GV (germinal vesicle stage) oocytes deficient in the heterochromatin protein 1β (HP1β), also known as the Chromobox Protein Homolog 1 (CBX1), and completely abrogates in oocytes deficient of the methyltransferase SUV39h2, in which the PCH regions lack both H3K9me3 and HP1β [45]. After fertilization, DAXX is revealed in the pronuclei of zygotes, initially in close association with atypical nucleoli [45][51][52], also known as the so-called nucleolus precursor bodies (NPBs), which serve as major organizing structures for heterochromatin in mammalian zygotes and early embryos [53]. The experiments, when the atypical nucleoli were removed from immature oocytes (enucleolation), have shown that the components of the maternal “nucleolus” are critically important for the deposition of DAXX in zygotes [51]. The absence of maternal “nucleoli” affects the dynamics of the S phase, and DAXX is no longer detected in enucleolated zygotes, suggesting that DAXX is not stably associated with maternal DNA.
As in somatic cells, H3.3 deposition in mammalian oocytes and embryos involves DAXX in the complex with ATRX. Moreover, both these proteins could operate in different complexes and assemble on chromatin with different kinetics [45][54]. The ATRX protein is necessary to recruit DAXX to PCH during meiotic prophase I. In the absence of ATRX, the DAXX protein fails to associate with the PCH regions of oocytes at the GV stage, despite of these regions contain H3K9me3, a mark of repressed chromatin [50]. In contrast with somatic cells in which the association of DAXX with ATRX at the PCH regions exists only for a brief period at the S phase [25][40], the DAXX/ATRX complex remains in association with these heterochromatin regions while an oocyte grows [50].
After fertilization, а clear colocalization of DAXX and ATRX initially revealed already at stage PN0 in the maternal pronucleus (mPN) and at stage PN2 in the paternal pronucleus (pPN), i.e., before replication, but thereafter, the DAXX level reduces rapidly in maternal PHC [45]. Importantly, the character of functional interactions between DAXX and ATRX after fertilization is fundamentally different in mPNs and pPNs, indicating a known asymmetry of the pronuclei in mammalian zygotes [45]. In the mPN, DAXX colocalizes with ATRX in the PCH regions of decondensed chromosomes containing H3K9me3 and HP1β. However, in the pPN, these chromatin regions contain only DAXX but not ATRX. It was also demonstrable that DAXX is necessary for ATRX recruitment to the PCH regions in paternal but not maternal pronuclei. In contrast, ATRX is required in the mPN to recruit DAXX, as shown in experiments with ATRX-depleted oocytes [45].
In mouse early embryos, DAXX-mediated H3.3 deposition is required for chromosome stability. The DAXX protein was shown to regulate repression of the Polycomb Repressive Complex 1 (PRC1) target genes in oogenesis and early embryogenesis. However, PRC2 and H3K27me3 do not serve as key determinants of DAXX recruitment and function in mouse zygotes [45]. The role of DAXX in maintaining the genome integrity is also evident during the period of global DNA demethylation that occurs after fertilization [55] and is accompanied by an increase in the recruitment of the DAXX/ATRX complex to tandemly repeating sequences, including retrotransposons and telomeres [33]. Knockdown of DAXX/ATRX in cells with hypomethylated DNA increases the aberrant derepression of tandem repeat transcription and expands telomere dysfunction. The DAXX/ATRX complex also suppresses H3K9 tri-methylation during mouse embryogenesis, interacting with the methyltransferase SUV39h [33].
As an H3.3 chaperone, DAXX is probably involved in remodeling of pericentromeric heterochromatin—the main place where H3.3 is deposited in oocytes and embryos. It has been shown that the mutation K27R in the H3.3 molecule results in the aberrant accumulation of pericentromeric transcripts, HP1 mislocalization, dysfunctional chromosome segregation, and developmental arrest [56]. Together with ATRX, the DAXX protein is also involved in repression of telomeric sequences. A central role for DAXX in association of the DAXX/ATRX complex with telomere/subtelomere regions was confirmed in experiments using mouse ESCs knocked out for DNA methyltransferases [33]. Loss of ATRX has a minor effect on the telomeric localization of DAXX, but knockdown of DAXX severely compromised the ability of ATRX to localize to telomeres. As DAXX and ATRX are localized in the telomeric regions of embryonic chromosomes, it cannot be excluded that in embryogenesis, DAXX and ATRX are involved in the Alternative Lengthening of Telomeres (ALT), especially since mutations in the DAXX gene have been described in patients with telomerase-negative pancreatic neuroendocrine cancers [57].
References
- Iqbal Mahmud; Daiqing Liao; DAXX in cancer: phenomena, processes, mechanisms and regulation. Nucleic Acids Research 2019, 47, 7734-7752, 10.1093/nar/gkz634.
- Peter W. Lewis; Simon J. Elsaesser; Kyung-Min Noh; Sonja C. Stadler; C. David Allis; Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proceedings of the National Academy of Sciences 2010, 107, 14075-14080, 10.1073/pnas.1008850107.
- Paolo Salomoni; The PML-Interacting Protein DAXX: Histone Loading Gets into the Picture. Frontiers in Oncology 2013, 3, 152, 10.3389/fonc.2013.00152.
- Michael A. Dyer; Zulekha A. Qadeer; David Valle-Garcia; Emily Bernstein; ATRX and DAXX: Mechanisms and Mutations. Cold Spring Harbor Perspectives in Medicine 2017, 7, a026567, 10.1101/cshperspect.a026567.
- Salvatore Fioriniello; Domenico Marano; Francesca Fiorillo; Maurizio D’Esposito; Floriana Della Ragione; Epigenetic Factors that Control Pericentric Heterochromatin Organization in Mammals. Genes 2020, 11, 595, 10.3390/genes11060595.
- Irina Bogolyubova; Dmitry Bogolyubov; DAXX Is a Crucial Factor for Proper Development of Mammalian Oocytes and Early Embryos. International Journal of Molecular Sciences 2021, 22, 1313, 10.3390/ijms22031313.
- Bogolyubova, I.O.; Bogolyubov, D.S. Oocyte nuclear structure during mammalian oogenesis. In Recent Advances in Germ Cells Research; Perrotte, A., Ed.; Nova Biomedical: New York, NY, USA, 2013; pp. 105–132.
- Irina Bogolyubova; Dmitry Bogolyubov; Heterochromatin Morphodynamics in Late Oogenesis and Early Embryogenesis of Mammals. Cells 2020, 9, 1497, 10.3390/cells9061497.
- Chuan-Wei Jang; Yoichiro Shibata; Joshua Starmer; Della Yee; Terry Magnuson; Histone H3.3 maintains genome integrity during mammalian development. Genes & Development 2015, 29, 1377-1392, 10.1101/gad.264150.115.
- Xiaolu Yang; Roya Khosravi-Far; Howard Y Chang; David Baltimore; Daxx, a Novel Fas-Binding Protein That Activates JNK and Apoptosis. Cell 1997, 89, 1067-1076, 10.1016/s0092-8674(00)80294-9.
- Howard Y. Chang; Hideki Nishitoh; Xiaolu Yang; Hidenori Ichijo; David Baltimore; Activation of Apoptosis Signal-Regulating Kinase 1 (ASK1) by the Adapter Protein Daxx. Science 1998, 281, 1860-1863, 10.1126/science.281.5384.1860.
- Seiji Torii; David A. Egan; Ronald A. Evans; John C. Reed; Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs). The EMBO Journal 1999, 18, 6037-6049, 10.1093/emboj/18.21.6037.
- Marianthi Kiriakidou; Deborah A. Driscoll; Jesus M. Lopez-Guisa; Jerome F. Strauss; Cloning and Expression of Primate Daxx cDNAs and Mapping of the Human Gene to Chromosome 6p21.3 in the MHC Region. DNA and Cell Biology 1997, 16, 1289-1298, 10.1089/dna.1997.16.1289.
- Eric Escobar-Cabrera; Desmond K.W. Lau; Serena Giovinazzi; Alexander M. Ishov; Lawrence P. McIntosh; Structural Characterization of the DAXX N-Terminal Helical Bundle Domain and Its Complex with Rassf1C. Structure 2010, 18, 1642-1653, 10.1016/j.str.2010.09.016.
- Simon J. Elsässer; Hongda Huang; Peter W. Lewis; Jason W. Chin; C. David Allis; Dinshaw J. Patel; DAXX envelops a histone H3.3–H4 dimer for H3.3-specific recognition. Nature 2012, 491, 560-565, 10.1038/nature11608.
- Chao-Pei Liu; Chaoyang Xiong; Mingzhu Wang; Zhouliang Yu; Na Yang; Ping Chen; Zhiguo Zhang; Guohong Li; Rui-Ming Xu; Structure of the variant histone H3.3–H4 heterodimer in complex with its chaperone DAXX. Nature Structural & Molecular Biology 2012, 19, 1287-1292, 10.1038/nsmb.2439.
- Aleixo Santiago; Adam C. Godsey; Jamil Hossain; Lisa Y. Zhao; Daiqing Liao; Identification of two independent SUMO-interacting motifs in Daxx: Evolutionary conservation from Drosophila to humans and their biochemical functions. Cell Cycle 2009, 8, 76-87, 10.4161/cc.8.1.7493.
- Xiaoman Wang; Yiyue Zhao; Jian Zhang; Yong Chen; Structural basis for DAXX interaction with ATRX. Protein & Cell 2017, 8, 767-771, 10.1007/s13238-017-0462-y.
- Jan Svadlenka; Jan Brazina; Hana Hanzlikova; Lukas Cermak; Ladislav Andera; Multifunctional adaptor protein Daxx interacts with chromatin-remodelling ATPase Brg1. Biochemistry and Biophysics Reports 2016, 5, 246-252, 10.1016/j.bbrep.2015.12.012.
- Alexander M. Ishov; Alexey G. Sotnikov; Dmitri Negorev; Olga V. Vladimirova; Norma Neff; Tetsu Kamitani; Edward T.H. Yeh; Jerome F. Strauss; Gerd G. Maul; Pml Is Critical for Nd10 Formation and Recruits the Pml-Interacting Protein Daxx to This Nuclear Structure When Modified by Sumo-1. Journal of Cell Biology 1999, 147, 221-234, 10.1083/jcb.147.2.221.
- A F Pluta; W C Earnshaw; I G Goldberg; Interphase-specific association of intrinsic centromere protein CENP-C with HDaxx, a death domain-binding protein implicated in Fas-mediated cell death.. Journal of Cell Science 1998, 111, 2029–2041.
- Jamie E. DeNizio; Simon J. Elsässer; Ben E. Black; DAXX co-folds with H3.3/H4 using high local stability conferred by the H3.3 variant recognition residues. Nucleic Acids Research 2014, 42, 4318-4331, 10.1093/nar/gku090.
- Valérie Lallemand-Breitenbach; Hugues De Thé; PML Nuclear Bodies. Cold Spring Harbor Perspectives in Biology 2010, 2, a000661, 10.1101/cshperspect.a000661.
- Hui Li; Christopher Leo; Jiang Zhu; Xiaoyang Wu; Jennifer O'neil; Eun-Ju Park; J. Don Chen; Sequestration and Inhibition of Daxx-Mediated Transcriptional Repression by PML. Molecular and Cellular Biology 2000, 20, 1784-1796, 10.1128/mcb.20.5.1784-1796.2000.
- Yutong Xue; Richard Gibbons; Zhijiang Yan; Dafeng Yang; Tarra L. McDowell; Salvatore Sechi; Jun Qin; Sharleen Zhou; Doug Higgs; Weidong Wang; et al. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proceedings of the National Academy of Sciences 2003, 100, 10635-10640, 10.1073/pnas.1937626100.
- Kami Ahmad; Steven Henikoff; The Histone Variant H3.3 Marks Active Chromatin by Replication-Independent Nucleosome Assembly. Molecular Cell 2002, 9, 1191-1200, 10.1016/s1097-2765(02)00542-7.
- Hideaki Tagami; Dominique Ray-Gallet; Geneviève Almouzni; Yoshihiro Nakatani; Histone H3.1 and H3.3 Complexes Mediate Nucleosome Assembly Pathways Dependent or Independent of DNA Synthesis. Cell 2004, 116, 51-61, 10.1016/s0092-8674(03)01064-x.
- Sandra B. Hake; Benjamin A. Garcia; Monika Kauer; Stephen P. Baker; Jeffrey Shabanowitz; Donald F. Hunt; C. David Allis; Serine 31 phosphorylation of histone variant H3.3 is specific to regions bordering centromeres in metaphase chromosomes. Proceedings of the National Academy of Sciences 2005, 102, 6344-6349, 10.1073/pnas.0502413102.
- Pascal Drané; Khalid Ouararhni; Arnaud Depaux; Muhammad Shuaib; Ali Hamiche; The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes & Development 2010, 24, 1253-1265, 10.1101/gad.566910.
- Aaron D. Goldberg; Laura A. Banaszynski; Kyung-Min Noh; Peter W. Lewis; Simon J. Elsaesser; Sonja Stadler; Scott Dewell; Martin Law; Xingyi Guo; Xuan Li; et al.Duancheng WenAriane ChapgierRussell C. DeKelverJeffrey C. MillerYa-Li LeeElizabeth A. BoydstonMichael C. HolmesPhilip D. GregoryJohn M. GreallyShahin RafiiChingwen YangPeter J. ScamblerDavid GarrickRichard J. GibbonsDouglas R. HiggsIleana M. CristeaFyodor D. UrnovDeyou ZhengC. David Allis Distinct Factors Control Histone Variant H3.3 Localization at Specific Genomic Regions. Cell 2010, 140, 678-691, 10.1016/j.cell.2010.01.003.
- Lee H. Wong; James D. McGhie; Marcus Sim; Melissa A. Anderson; Soyeon Ahn; Ross D. Hannan; Amee J. George; Kylie A. Morgan; Jeffrey R. Mann; K.H. Andy Choo; et al. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Research 2010, 20, 351-360, 10.1101/gr.101477.109.
- Simon J. Elsässer; Kyung-Min Noh; Nichole Diaz; C. David Allis; Laura A. Banaszynski; Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 2015, 522, 240-244, 10.1038/nature14345.
- Quanyuan He; Hyeung Kim; Rui Huang; Weisi Lu; Mengfan Tang; Fengtao Shi; Dong Yang; Xiya Zhang; Junjiu Huang; Dan Liu; et al.Zhou Songyang The Daxx/Atrx Complex Protects Tandem Repetitive Elements during DNA Hypomethylation by Promoting H3K9 Trimethylation. Cell Stem Cell 2015, 17, 273-286, 10.1016/j.stem.2015.07.022.
- Maheshi Udugama; Fiona T. M. Chang; F. Lyn Chan; Michelle C. Tang; Hilda A. Pickett; James D. R. McGhie; Lynne Mayne; Philippe Collas; Jeffrey R. Mann; Lee H. Wong; et al. Histone variant H3.3 provides the heterochromatic H3 lysine 9 tri-methylation mark at telomeres.. Nucleic Acids Research 2015, 43, 10227-10237, 10.1093/nar/gkv847.
- Hsiao P.J. Voon; Jim R. Hughes; Christina Rode; Inti A. De La Rosa-Velázquez; Thomas Jenuwein; Robert Feil; Douglas R. Higgs; Richard J. Gibbons; ATRX Plays a Key Role in Maintaining Silencing at Interstitial Heterochromatic Loci and Imprinted Genes. Cell Reports 2015, 11, 405-418, 10.1016/j.celrep.2015.03.036.
- Viacheslav M. Morozov; Serena Giovinazzi; Alexander M. Ishov; CENP-B protects centromere chromatin integrity by facilitating histone deposition via the H3.3-specific chaperone Daxx. Epigenetics & Chromatin 2017, 10, 63, 10.1186/s13072-017-0164-y.
- Prashanth Krishna Shastrula; Isabel Sierra; Zhong Deng; Frederick Keeney; James E. Hayden; Paul M. Lieberman; Susan M. Janicki; PML is recruited to heterochromatin during S phase and represses DAXX-mediated histone H3.3 chromatin assembly. Journal of Cell Science 2019, 132, jcs220970, 10.1242/jcs.220970.
- Erwan Delbarre; Kristina Ivanauskiene; Jane Spirkoski; Akshay Shah; Kristin Vekterud; Jan Øivind Moskaug; Stig Ove Bøe; Lee H. Wong; Thomas Küntziger; Philippe Collas; et al. PML protein organizes heterochromatin domains where it regulates histone H3.3 deposition by ATRX/DAXX. Genome Research 2017, 27, 913-921, 10.1101/gr.215830.116.
- Lindsy M. Rapkin; Kashif Ahmed; Stanimir Dulev; Ren Li; Hiroshi Kimura; Alexander M. Ishov; David P. Bazett-Jones; The histone chaperone DAXX maintains the structural organization of heterochromatin domains. Epigenetics & Chromatin 2015, 8, 44, 10.1186/s13072-015-0036-2.
- Alexander M. Ishov; Olga V. Vladimirova; Gerd G. Maul; Heterochromatin and ND10 are cell-cycle regulated and phosphorylation-dependent alternate nuclear sites of the transcription repressor Daxx and SWI/SNF protein ATRX. Journal of Cell Science 2004, 117, 3807-3820, 10.1242/jcs.01230.
- Serena Giovinazzi; Cory R. Lindsay; Viacheslav M. Morozov; Eric Escobar-Cabrera; Matthew K. Summers; Hyo Sook Han; Lawrence P. McIntosh; Alexander M. Ishov; Regulation of mitosis and taxane response by Daxx and Rassf1. Oncogene 2011, 31, 13-26, 10.1038/onc.2011.211.
- S Giovinazzi; V M Morozov; M K Summers; W C Reinhold; A M Ishov; USP7 and Daxx regulate mitosis progression and taxane sensitivity by affecting stability of Aurora-A kinase. Cell Death & Differentiation 2013, 20, 721-731, 10.1038/cdd.2012.169.
- Viacheslav M. Morozov; Ekaterina V. Gavrilova; Vasily V. Ogryzko; Alexander M. Ishov; Dualistic function of Daxx at centromeric and pericentromeric heterochromatin in normal and stress conditions. Nucleus 2012, 3, 276-285, 10.4161/nucl.20180.
- Jennifer S. Michaelson; Debra Bader; Frank Kuo; Christine Kozak; Philip Leder; Loss of Daxx, a promiscuously interacting protein, results in extensive apoptosis in early mouse development. Genes & Development 1999, 13, 1918-1923, 10.1101/gad.13.15.1918.
- Zichuan Liu; Mathieu Tardat; Mark E Gill; Helene Royo; Raphael Thierry; Evgeniy A Ozonov; Antoine Hfm Peters; SUMO ylated PRC 1 controls histone H3.3 deposition and genome integrity of embryonic heterochromatin. The EMBO Journal 2020, 39, e103697, 10.15252/embj.2019103697.
- Duancheng Wen; Laura A. Banaszynski; Ying Liu; Fuqiang Geng; Kyung-Min Noh; Jenny Xiang; Olivier Elemento; Zev Rosenwaks; C. David Allis; Shahin Rafii; et al. Histone variant H3.3 is an essential maternal factor for oocyte reprogramming. Proceedings of the National Academy of Sciences 2014, 111, 7325-7330, 10.1073/pnas.1406389111.
- Tomohiko Akiyama; Osamu Suzuki; Junichiro Matsuda; Fugaku Aoki; Dynamic Replacement of Histone H3 Variants Reprograms Epigenetic Marks in Early Mouse Embryos. PLOS Genetics 2011, 7, e1002279, 10.1371/journal.pgen.1002279.
- Sung-Joon Park; Makiko Komata; Fukashi Inoue; Kaori Yamada; Kenta Nakai; Miho Ohsugi; Katsuhiko Shirahige; Inferring the choreography of parental genomes during fertilization from ultralarge-scale whole-transcriptome analysis. Genes & Development 2013, 27, 2736-2748, 10.1101/gad.227926.113.
- Buhe Nashun; Peter W.S. Hill; Sebastien A. Smallwood; Gopuraja Dharmalingam; Rachel Amouroux; Stephen J. Clark; Vineet Sharma; Elodie Ndjetehe; Pawel Pelczar; Richard J. Festenstein; et al.Gavin KelseyPetra Hajkova Continuous Histone Replacement by Hira Is Essential for Normal Transcriptional Regulation and De Novo DNA Methylation during Mouse Oogenesis. Molecular Cell 2015, 60, 611-625, 10.1016/j.molcel.2015.10.010.
- Claudia Baumann; Maria M. Viveiros; Rabindranath De La Fuente; Loss of Maternal ATRX Results in Centromere Instability and Aneuploidy in the Mammalian Oocyte and Pre-Implantation Embryo. PLOS Genetics 2010, 6, e1001137, 10.1371/journal.pgen.1001137.
- H. Fulka; A. Langerova; The maternal nucleolus plays a key role in centromere satellite maintenance during the oocyte to embryo transition. Development 2014, 141, 1694-1704, 10.1242/dev.105940.
- Irina O. Bogolyubova; Zhuldyz K. Sailau; Dmitry S. Bogolyubov; The dynamics of DAXX protein distribution in the nucleus of mouse early embryos. Acta Histochemica 2019, 121, 522-529, 10.1016/j.acthis.2019.04.007.
- Josef Jr. Fulka; Michal Benc; Pasqualino Loi; Alena Langerova; Helena Fulka; Function of atypical mammalian oocyte/zygote nucleoli and its implications for reproductive biology and medicine. The International Journal of Developmental Biology 2019, 63, 105-112, 10.1387/ijdb.180329jf.
- Rabindranath De La Fuente; Claudia Baumann; Maria M. Viveiros; ATRX contributes to epigenetic asymmetry and silencing of major satellite transcripts in the maternal genome of the mouse embryo. Development 2015, 142, 1806-1817, 10.1242/dev.118927.
- Fátima Santos; Wendy Dean; Epigenetic reprogramming during early development in mammals. Reproduction 2004, 127, 643-651, 10.1530/rep.1.00221.
- Angèle Santenard; Céline Ziegler-Birling; Marc Koch; Làszlò Tora; Andrew J. Bannister; Maria-Elena Torres-Padilla; Heterochromatin formation in the mouse embryo requires critical residues of the histone variant H3.3. Nature 2010, 12, 853-862, 10.1038/ncb2089.
- Yuchen Jiao; Chanjuan Shi; Barish H. Edil; Roeland F. De Wilde; David S. Klimstra; Anirban Maitra; Richard D. Schulick; Laura H. Tang; Christopher L. Wolfgang; Michael A. Choti; et al.Victor E. VelculescuLuis A. DiazBert VogelsteinKenneth W. KinzlerRalph H. HrubanNickolas Papadopoulos DAXX/ATRX, MEN1, and mTOR Pathway Genes Are Frequently Altered in Pancreatic Neuroendocrine Tumors. Science 2011, 331, 1199-1203, 10.1126/science.1200609.
- Yuchen Jiao; Chanjuan Shi; Barish H. Edil; Roeland F. De Wilde; David S. Klimstra; Anirban Maitra; Richard D. Schulick; Laura H. Tang; Christopher L. Wolfgang; Michael A. Choti; et al.Victor E. VelculescuLuis A. DiazBert VogelsteinKenneth W. KinzlerRalph H. HrubanNickolas Papadopoulos DAXX/ATRX, MEN1, and mTOR Pathway Genes Are Frequently Altered in Pancreatic Neuroendocrine Tumors. Science 2011, 331, 1199-1203, 10.1126/science.1200609.

