 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Eva Estebanez-Perpina | + 2929 word(s) | 2929 | 2021-02-25 04:44:11 | | | |
| 2 | Peter Tang | Meta information modification | 2929 | 2021-03-08 02:21:12 | | |
Video Upload Options
The Androgen Receptor (NR3C4, nuclear receptor subfamily 3, group C, gene 4) is a member of the steroid hormone group of nuclear receptors along with the oestrogen receptors (ERα and β isoforms, NR3A1 and NR2A2, respectively), glucocorticoid receptor (GR, NR3C1), progesterone receptor (PR, NR3C3) and mineralocorticoid receptor (MR, NR3C2).
1. Introduction
Prostate cancer (PCa) is the second most common cancer in men world-wide, with nearly 1.3 million new cases each year [1]. Over the last ten years this equates to nearly 12 million men being diagnosed with PCa. It is estimated that, in the USA, 1 in 41 men will die of PCa [2] (and over the next twenty years the incidence and death rate are predicted to nearly double [3]. Interestingly, while this increase is seen in most regions of the world, it is predicted to be highest in Latin America/Caribbean, Africa and Asia [3]. Other than genetic factors a number of risk factors have been proposed for the increase in PCa: diet and age, as a result of increased life expectancy, have been particularly highlighted. The real consequence behind these statistics is that increasing numbers of men will be living with this disease and there will be an increased burden on health providers and resources.
PCa is not a new disease and there are an increasing number of detailed analyses of archaeological samples that reveal evidence of metastatic PCa in the ancient world (Figure 1) [4][5][6][7]. The most compelling evidence comes from molecular and microscopy studies on the preserved remains of a Scythian king, discovered in Siberia and presumed to have died of PCa [7]. Schmidt-Schultz and co-workers reported on the presence of bone lesions consistent with invasive growth or enzymatic activity and strikingly showed increased levels of PSA (in complex with 1-antichymotrypsin) [7]. Apart from the technological achievement of this study, it is particularly noteworthy that the same changes associated with PCa in modern patients were identified in a 2700-year-old man.
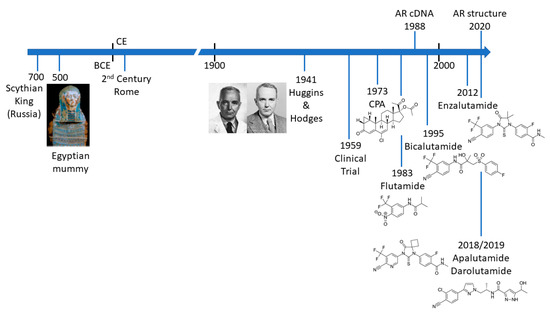
Figure 1. Timeline illustrating key events in the treatment of prostate cancer.
The Venetian physician and anatomist, Niccolò Massa (1495 to 1569) is credited with the first true description of the prostate gland in the 16th century. However, due to the location of the prostate gland, access to surgically excise tumours was seen as particularly challenging and from the mid-18th century to the early 20th century different approaches were trialled. Due to pioneering approaches and the skill of early surgeons such as Eugene Fuller (New York), Peter J. Freyer (UK), Hugh H. Young (Baltimore) and H. Kuchler (Germany) radical proctectomy has been a standard of care for over 100 years (reviewed in [8]). Continuing refinements to improve patient recovery and preserve quality of life include the development of laparoscopic and robotic surgery, as well as (controversially) focussed therapy in an attempt to avoid side-effects associated with surgery [9]. Of course, surgery is only a curative option in PCa confined to the prostate gland. Metastatic disease must be treated systemically and here the role of the androgen receptor (AR) becomes paramount.
2. Androgen Receptor—A Key Driver of PCa and Drug Target
The differentiation and development of the prostate is dependent upon the androgen dihydrotestosterone (DHT), which can be derived directly from testosterone in prostate cells or by an alternative pathway in the embryo [10][11][12]. In a seminal paper in 1941, Charles Huggins and Clarence V. Hodges demonstrated that prostate tumours and metastatic disease were also sensitive to the presence or absence of androgenic hormones (Figure 1) [13]. Thus, reduction of circulating testosterone, by removal of the testis or treatment with a synthetic oestrogen, stilbesterol, resulted in reduction in acid phosphatase, an early biomarker for PCa [13]. The significance and influence of this study for the treatment of advanced PCa cannot be overstated.
The “classical” model for androgen action, via the AR, is illustrated in Figure 2. The AR protein resides in the cytoplasm bound to chaperone complexes, but upon hormone binding to the AR with high affinity, the receptor–chaperone complex rearranges, an intramolecular interaction occurs between the amino-terminal and carboxy-terminal ends [14][15] and importin-α [16] is recruited to translocate the AR into the nucleus (Figure 2). In the nucleus, receptor dimers bind to sequence-specific androgen response elements (AREs) in the promoter and enhancer region of target genes, such as prostate-specific antigen (PSA) and transmembrane protease serine 2 (TMPRSS2). Once bound to the chromatin, AR recruits numerous coregulatory proteins to modulate transcription, leading to cell growth and survival responses [17][18][19]. At the genome level, the AR recruits members of the basal transcription machinery, for instance TATA-box-binding protein (TBP) and transcription factor IIF (TFIIF), and also other pivotal coregulators such as different members of the p160 family of coactivators and cAMP-response element-binding protein (CREB)-binding protein (CBP) (see [18][20][21][22]). However, this “simple” monomer–dimer equilibrium transition model is being challenged by ongoing research, and important questions remain regarding the possible additional role of chaperone proteins [23][24][25][26], the cellular location of dimerization [15][27][28] and the influence of response element architecture on receptor activity. Furthermore, recent studies also show that besides monomer and dimer forms, AR exists in tetramers and higher multimer oligomers even in the absence of hormone in the nucleus [28][29][30].
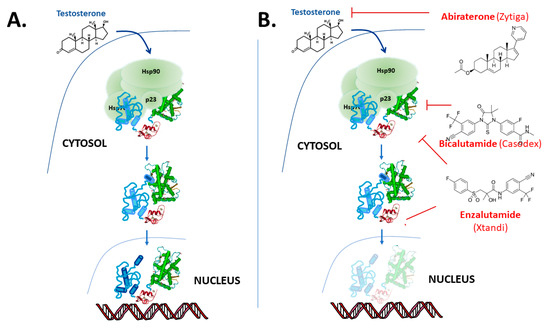
Figure 2. Overview of androgen receptor mechanism of action. (A) In the “classical” model the AR binds to testosterone and in tissues such as the prostate preferentially to the more potent metabolite 5α-dihydrotestosterone (DHT) and dissociates from molecular chaperones and translocates to the nucleus where it binds to DNA response elements and up- or down-regulates target gene expression. However, the details of a number of these steps in this model remain subject to debate and on-going research. (B) The action of the drugs abiraterone, bicalutamide and enzalutamide used in the treatment of PCa.
3. Androgen Receptor Structure and Function
The AR (NR3C4, nuclear receptor subfamily 3, group C, gene 4) is a member of the steroid hormone group of nuclear receptors along with the oestrogen receptors (ERα and β isoforms, NR3A1 and NR2A2, respectively), glucocorticoid receptor (GR, NR3C1), progesterone receptor (PR, NR3C3) and mineralocorticoid receptor (MR, NR3C2). The AR gene is present on the long arm on the X chromosome (Xq11-12) and contains eight exons interrupted by introns of varying lengths (0.7–2.6 kb) and codes for a protein of approximately 920 amino acids. There is variation in overall length due to polymorphic amino acid repeat regions within the N-terminal region. One of these, a polyglutamine repeat, is associated with the neuronal degenerative disorder Spinal and Bulbar Muscular Atrophy (SBMA) when the glutamines number greater than 38 (normal range is 9–34 in Caucasian populations) [31]. A predominant mRNA transcript of 10–11 kb and smaller species, between 6 and 8.5 kb, have been described in human breast and prostate tissue and cell lines [32][33]. The AR protein is composed of several functional domains: the intrinsically disordered N-terminal domain (NTD), the DNA binding domain (DBD) and the ligand binding domain (LBD) (Figure 3). The NTD is coded by exon 1, the DBD by exons 2 and 3, while exons 4 to 8 encode both the hinge and LBD modules [34]. We herein provide an overview of detailed AR structure and activity, its actions in PCa, and how the combination of structural information and functional screenings has been used for drug discovery of AR modulators.

Figure 3. Functional and structural domains of the androgen receptor. The full-length androgen receptor (AR-FL) has a variable number of amino acids due to highly polymorphic glutamine (Q) and glycine (G) repeats in the amino-terminal domain (NTD). Splice variants (AR-Vs) lacking the ligand-binding domain (LBD), but retaining the DNA-binding domain (DBD) and NTD, are thought to emerge in PCa as a consequence of androgen ablation therapy. Available structural information for isolated ligand-binding (PDB 1I37) and DNA-binding (PBD 1R4I) domains is shown above the schematic of the AR-FL. Note—in the text the number of amino acids and point mutations is based on AR-FL of 920 amino acids.
3.1. The N-Terminal Domain
The AR-NTD makes up more than 50% of the receptor protein and shows little or no sequence homology with even its closest relatives, the GR and PR [34]. Importantly, the NTD contains activation function (AF) 1 and is essential for receptor-dependent transcriptional activation. AR-AF1 is modular in structure and function with two overlapping sub-domains: TAU1 (amino acids 103 to 372) and TAU5 (amino acids 362 to 538) [22][35][36][37][38].
In contrast to the rest of the protein the AR-NTD is intrinsically disordered and demonstrates a high degree of structural plasticity (reviewed in [39][40]), with the AF1 region shown to have a “collapsed disordered” conformation [41]. Regions of α-helical structure have been mapped by secondary structure predictions, mutagenesis and circular dichroism [37][41][42][43] and more recently by high-resolution NMR spectroscopy [21][22][44][45] (Figure 3). Intrinsically disordered protein domains allow for the coupling of protein binding and folding, which facilitates selective interactions with multiple binding partners (reviewed in [40]). Intrinsic disorder of the GR NTD has been shown to mediate allosteric regulation of receptor function and coupling transcriptional activation or repression activity with the receptor DBD [46]. Intriguingly, in the recent cryo-EM structure of the AR-FL bound to DNA the NTD was observed to form an asymmetric ring fold surrounding the DBD and LBD, which created surfaces for co-regulatory protein binding [18]. This visualisation of the AR-NTD suggests increased structural stability in the context of the DNA bound dimer complex and/or the antibody binding used to locate the domain in the structure.
In the case of the AR the presence of the NTD has also been shown to modulate DNA binding affinity for different response elements [47]. There is also considerable evidence showing that maximal activity of the AR requires an intramolecular interaction between the NTD and the ligand-binding domain, termed the N/C interaction, that occurs when ligands bind [14][48].
3.2. The DNA Binding Domain
The DNA-binding domain (DBD) is the most well-conserved region between different steroid receptors and is defined by nine conserved cysteine residues. The AR-DBD has a helical-globular structure (Figure 3) in which three sub-regions can be distinguished: two zinc fingers and a more loosely structured carboxy terminal extension (CTE) [49][50]. The DBD fold is stabilized by the coordination of two zinc ions by eight of the conserved cysteines [49]. The first zinc finger is important for recognition and binding of DNA response elements: three amino acid residues form the “P-box” (Gly578, Ser579Val582) as part of the DNA recognition helix. Five amino acids in the second zinc cluster form the “D-box” (Ala597-Ser-Arg-Asn-Asp601), involved in receptor dimerization. The P-box residues are conserved in the GR, PR and MR leading to shared response elements (often termed hormone response elements or HREs) and potentially overlapping gene signatures. However, the AR-DBD dimerisation and the CTE are thought to contribute to specific binding to selective DNA sequences, termed androgen response elements (AREs) ([49][50] and references therein). Furthermore, a relaxed stringency in the DNA sequence has been observed to be important for chromatin binding of the AR to selective response elements [51]. These different types of AR binding sites have been shown to be important for mediating normal physiology [51][52]. They may also play a role in resistance to antiandrogen therapy through GR activity [53] and have been suggested to underpin a differential response of PCa cells to a chemotherapy agent [54].
In a mouse model, termed “specificity affecting AR knock-in” (SPARKI) it was found that loss of AR binding to such selective AREs resulted in infertility (in male animals) due to impaired sperm maturation in the epididymis [52]. More recently, Robins and co-workers reported that low concentrations of doxorubicin preferentially inhibited AR target genes associated with HREs, while genes driven by selective AREs were upregulated under these conditions [54]. In ChIP-seq studies the increased binding of the AR, in the presence of low concentrations of doxorubicin, appeared to involve a redistribution of the receptor and cooperativity with other transcription factors, notably NKX3.1, HOXB13 and the pioneer factor FOXA1. However, strikingly, no identifiable HRE or ARE element was found at these sites [54]. Taken together these studies emphasize the importance of both DNA response element sequence and synergy with other DNA-binding proteins for both tissue selective gene expression and changes in the AR gene signature seen in PCa progression.
3.3. The Ligand Binding Domain
The onset, development and progression of PCa depends on androgenic hormone activation of the receptor and from the early work of Huggins and Hodges the AR is recognized as a crucial drug target in the fight against this cancer. First, PCa is treated by depriving tumours of androgens such as DHT and testosterone or blocking their actions by impeding their direct binding to the ligand binding pocket (LBP) on the LBD (Figure 3). The compounds impeding or blocking the binding of androgenic hormones are called anti-androgens (Figure 4). However, the effect of this type of antagonist treatment is transient, as universally patients relapse within a few years after developing a castration-resistant form of the disease, which is usually due to increased levels of AR expression or point-mutations that cause the AR to be resistant to anti-androgens, see below.
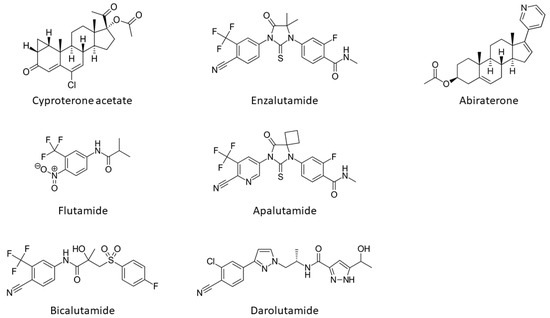
Figure 4. Structures of androgen receptor antagonists and the CYP17A1 inhibitor, abiraterone.
The determination of the atomic three-dimensional crystal structures provided a framework for understanding AR function and revealed detailed molecular determinants for the recognition and binding of cognate natural ligands, allowing rational drug design for the treatment of PCa. First of all, the pioneer structures revealed the folding of the LBD of AR and exhibited its overall conserved typical/canonical nuclear receptor fold. In particular, AR LBD structure contains nine α-helices, two 310 helices, and four short β-strands assembled in two anti-parallel β-sheets (Figure 3). The helices are arranged in an alpha-helical sandwich; in this particular receptor the helices H4, H5, H10-11 are disposed contiguously, and H12 is folded against the body of the domain, exhibiting a completely formed coactivator binding groove also known as coactivator binding pocket or AF-2. AR does not contain a helix 2, but a long loop linking H1 with H3. Furthermore, AR and the other oxosteroid receptors feature an amino acid stretch right after the last helix H12, which is called the F-domain, a structural element that hampers the LBD domain from dimerizing using the canonical LDB dimer architecture [30]. It is also worth noting that the AR uses a novel dimerization interface involving residues in helix 5 [27].
These early models also allowed the characterization of the cocooned ligand binding pocket (LBP) and detailed atomic information on how the hormones or analogous molecules were nested in the pocket. This initial information allowed more predictive models of designed compound binding and fuelled intense drug discovery programs to develop more selective competitive AR modulators (SARMs). Since these first structures of the LBD [55], structure-based drug discoveries in academic and pharmaceutical settings have continuously been reported. Many studies have focused on providing new insights into the mechanisms of AR-targeted compounds and action in PCa but most importantly how to design more potent and selective anti-androgens with fewer side effects that may bypass resistance.
The LBP of AR exhibits numerous residues (Figure 5), which form important contacts with the natural hormone or the ligand metribolone (R1881). A total of twenty residues interact with the agonist in the LBP. Most of the residues involved are hydrophobic and are responsible for the interaction with the steroid scaffold of the agonists. The remaining residues are polar and engage in hydrogen bonds with the polar atoms of the ligand; conserved water molecules have also been described trapped in the internal cavity [56].
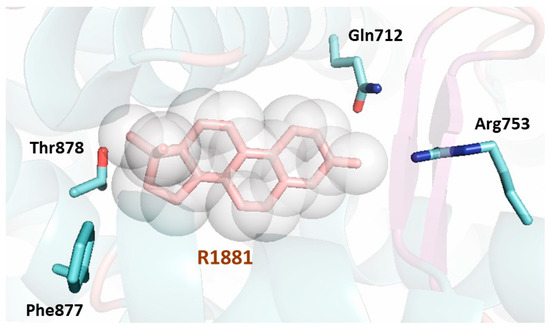
Figure 5. Cartoon representation of the first AR LBD crystal structure solved (PDB [Protein Data Bank] code 1E3G) [55]. The AR LBD secondary structure is shown depicting the helices, loops, and β-sheets surrounding the ligand (shown as a stick model in pink surrounded by a sphere representation). The LBP of the AR LBD is formed by twenty residues lining the bound ligand. Three residues make hydrogen bonds with the ligand (Gln712, Arg753 and Thr878) and are shown as sticks in blue showing their lateral chains. The residue Phe877, also shown as a stick representation in blue, has been found to be linked to enzalutamide resistance. All the residues lining the pocket extracted from the PISA server (https://www.ebi.ac.uk/pdbe/pisa/) are: Leu702, Leu705, Asn706, Leu708, Gly709, Gln712, Trp742, Met743, Met746, Val747, Met750, Arg753, Phe765, Met781, Met788, Leu 874, Phe877, Leu881, Met896 and Ile900.
Several point-mutations in the LBP (Figure 5) have been described to be associated with PCa or androgen insensitivity syndromes and the crystal structures of agonist-bound AR LBD provided a structural basis to explain their impact on the structure–function relationship underlying the receptor functionality under (patho)physiological conditions. Most importantly, the difference in some of the residues that form the LBP is what confers ligand specificity. The structures provided information on how mutations linked to PCa clustered near the position 17 beta hydroxyl group of the ligand, while mutations associated with androgen insensitivity syndromes clustered around other parts of the bound ligand. There are also some residues that are found mutated in both diseases, and mutations identified are summarised in the AR Mutations Database (http://androgendb.mcgill.ca) [57].
Mutations in the LBP have been shown to underlie a switch in activity of some therapeutic antagonists to have agonist activity. These point mutations encode residue substitutions that increase the agonist activity of the receptor because it no longer recognizes clinical anti-androgens as antagonists, and the receptor continues being activated. Several mutations inside the LBP (e.g., T878A, W742L, F877L) have been found in advanced tumours and observed to result in the acquirement of agonist activity of anti-androgens. Most of these mutations expand the LBP cavity by concrete structural rearrangements in the surrounding helices conforming the walls of the pocket. So AR LBP, as has been shown for other related receptors, exhibits certain structural plasticity and flexibility in its LBP that allows the protein to rearrange—induced by the action of designed ligands and/or point mutations that are mostly treatment-induced [58].
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Noone, A.M. Cancer Statistics Review 1975–2015; National Cancer Institute: Bethesda, MD, USA, 2017.
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89.
- Ghabili, K.; Tosoian, J.J.; Schaeffer, E.M.; Pavlovich, C.P.; Golzari, S.E.J.; Khajir, G.; Andreas, D.; Benzon, B.; Vuica-Ross, M.; Ross, A.E. The History of Prostate Cancer from Antiquity: Review of Paleopathological Studies. Urology 2016, 97, 8–12.
- Minozzi, S.; Lunardini, A.; Caldarini, C.; Caramella, D.; Fornaciari, G.; Catalano, P.; Giuffra, V. Metastatic Prostate Carcinoma from Imperial Rome (1st to 2nd Centuries AD). Pathobiology 2018, 85, 289–299.
- Prates, C.; Sousa, S.; Oliveira, C.; Ikram, S. Prostate metastatic bone cancer in an Egyptian Ptolemaic mummy, a proposed radiological diagnosis. Int. J. Paleopathol. 2011, 1, 98–103.
- Schultz, M.; Parzinger, H.; Posdnjakov, D.V.; Chikisheva, T.A.; Schmidt-Schultz, T.H. Oldest known case of metastasizing prostate carcinoma diagnosed in the skeleton of a 2,700-year-old Scythian king from Arzhan (Siberia, Russia). Int. J. Cancer 2007, 121, 2591–2595.
- Sriprasad, S.; Feneley, M.R.; Thompson, P.M. History of prostate cancer treatment. Surg. Oncol. 2009, 18, 185–191.
- Guillaumier, S.; Peters, M.; Arya, M.; Afzal, N.; Charman, S.; Dudderidge, T.; Hosking-Jervis, F.; Hindley, R.G.; Lewi, H.; McCartan, N.; et al. A Multicentre Study of 5-year Outcomes Following Focal Therapy in Treating Clinically Significant Nonmetastatic Prostate Cancer. Eur. Urol. 2018, 74, 422–429.
- Fukami, M.; Homma, K.; Hasegawa, T.; Ogata, T. Backdoor pathway for dihydrotestosterone biosynthesis: Implications for normal and abnormal human sex development. Dev. Dyn. 2012, 242, 320–329.
- O’Shaughnessy, P.J.; Antignac, J.P.; Le Bizec, B.; Morvan, M.-L.; Svechnikov, K.; Söder, O.; Savchuk, I.; Monteiro, A.; Soffientini, U.; Johnston, Z.C.; et al. Alternative (backdoor) androgen production and masculinization in the human fetus. PLoS Biol. 2019, 17, e3000002.
- Wilson, J.D.; Griffin, J.E.; Russell, D.W. Steroid 5 Alpha-Reductase 2 Deficiency. Endocr. Rev. 1993, 14, 577–593.
- Huggins, C.; Hodges, C.V. Studies on Prostatic Cancer: I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. J. Urol. 2002, 168, 9–12.
- He, B.; Gampe, R.T.; Kole, A.J.; Hnat, A.T.; Stanley, T.B.; An, G.; Stewart, E.L.; Kalman, R.I.; Minges, J.T.; Wilson, E.M. Structural Basis for Androgen Receptor Interdomain and Coactivator Interactions Suggests a Transition in Nuclear Receptor Activation Function Dominance. Mol. Cell 2004, 16, 425–438.
- Van Royen, M.E.; Cunha, S.M.; Brink, M.C.; Mattern, K.A.; Nigg, A.L.; Dubbink, H.J.; Verschure, P.J.; Trapman, J.; Houtsmuller, A.B. Compartmentalization of androgen receptor protein–protein interactions in living cells. J. Cell Biol. 2007, 177, 63–72.
- Cutress, M.L.; Whitaker, H.C.; Mills, I.G.; Stewart, M.; Neal, D.E. Structural Basis for the Nuclear Import of the Human An-drogen Receptor. J. Cell. Sci. 2008, 121, 957–968.
- Wang, Q.; Li, W.; Liu, X.S.; Carroll, J.S.; Jänne, O.A.; Keeton, E.K.; Chinnaiyan, A.M.; Pienta, K.J.; Brown, M. A Hierarchical Network of Transcription Factors Governs Androgen Receptor-Dependent Prostate Cancer Growth. Mol. Cell 2007, 27, 380–392.
- Yu, X.; Yi, P.; Hamilton, R.A.; Shen, H.; Chen, M.; Foulds, C.E.; Mancini, M.A.; Ludtke, S.J.; Wang, Z.; O’Malley, B.W. Structural Insights of Transcriptionally Active, Full-Length Androgen Receptor Coactivator Complexes. Mol. Cell 2020, 79, 812–823.e4.
- Powell, S.M.; Christiaens, V.; Voulgaraki, D.; Waxman, J.; Claessens, F.; Bevan, C.L. Mechanisms of Androgen Receptor Sig-nalling Via Steroid Receptor Coactivator-1 in Prostate. Endocr. Relat. Cancer 2004, 11, 117–130.
- Bevan, C.L.; Hoare, S.; Claessens, F.; Heery, D.M.; Parker, M.G. The AF1 and AF2 Domains of the Androgen Receptor Inter-act with Distinct Regions of SRC1. Mol. Cell. Biol. 1999, 19, 8383–8392.
- De Mol, E.; Szulc, E.; di Sanza, C.; Marttnez-Cristtbal, P.; Bertoncini, C.W.; Fenwick, R.B.; Frigoll-Vivas, M.; Masín, M.; Hunter, I.; Buzzn, V.; et al. Regulation of Androgen Receptor Activity by Transient Interactions of Its Transactivation Domain with General Transcription Regulators. SSRN Electron. J. 2018, 26, 145.
- De Moll, E.; Fenwick, R.B.; Phang, C.T.W.; Buzón, V.; Szulc, E.; de la Fuente, A.; Escobedo, A.; García, J.; Bertoncini, C.W.; Estébanez-Perpiñá, E.; et al. EPI-001, A Compound Active against Castration-Resistant Prostate Cancer, Targets Transactivation Unit 5 of the Androgen Receptor. ACS Chem. Biol. 2016, 11, 2499–2505.
- Cano, L.Q.; Lavery, D.N.; Bevan, C.L. Mini-review: Foldosome regulation of androgen receptor action in prostate cancer. Mol. Cell. Endocrinol. 2013, 369, 52–62.
- Reebye, V.; Querol-Cano, L.; Lavery, D.N.; Brooke, G.N.; Powell, S.M.; Chotai, D.; Walker, M.M.; Whitaker, H.C.; Wait, R.; Hurst, H.C.; et al. Role of the HSP90-Associated Cochaperone p23 in Enhancing Activity of the Androgen Receptor and Significance for Prostate Cancer. Mol. Endocrinol. 2012, 26, 1694–1706.
- Shatkina, L.; Mink, S.; Rogatsch, H.; Klocker, H.; Langer, G.; Nestl, A.; Cato, A.C. The Cochaperone Bag-1L Enhances Androgen Receptor Action Via Interaction with the NH2-Terminal Region of the Receptor. Mol. Cell. Biol. 2003, 23, 7189–7197.
- Yong, W.; Yang, Z.; Periyasamy, S.; Chen, H.; Yucel, S.; Li, W.; Lin, L.Y.; Wolf, I.M.; Cohn, M.J.; Baskin, L.S.; et al. Essential Role for Co-chaperone Fkbp52 but Not Fkbp51 in Androgen Receptor-mediated Signaling and Physiology. J. Biol. Chem. 2007, 282, 5026–5036.
- Nadal, M.; Prekovic, S.; Gallastegui, N.; Helsen, C.; Abella, M.; Zielinska, K.; Gay, M.; Vilaseca, M.; Taulès, M.; Houtsmuller, A.B.; et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat. Commun. 2017, 8, 14388.
- Presman, D.M.; Hager, G.L. More than meets the dimer: What is the quaternary structure of the glucocorticoid receptor? Transcription 2017, 8, 32–39.
- Presman, D.M.; Ganguly, S.; Schiltz, R.L.; Johnson, T.A.; Karpova, T.S.; Hager, G.L. DNA binding triggers tetramerization of the glucocorticoid receptor in live cells. Proc. Natl. Acad. Sci. USA 2016, 113, 8236–8241.
- Jimenez-Panizo, A.; Perez, P.; Rojas, A.M.; Fuentes-Prior, P.; Estebanez-Perpina, E. Non-Canonical Dimerization of the An-drogen Receptor and Other Nuclear Receptors: Implications for Human Disease. Endocr. Relat. Cancer 2019, 26, R479–R497.
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; E Harding, A.; Fischbeck, K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nat. Cell Biol. 1991, 352, 77–79.
- Faber, P.W.; van Rooij, H.C.; van der Korput, H.A.; Baarends, W.M.; Brinkmann, A.O.; Grootegoed, J.A.; Trapman, J. Characterization of the Human Androgen Receptor Transcription Unit. J. Biol. Chem. 1991, 266, 10743–10749.
- Tilley, W.D.; Wilson, C.M.; Marcelli, M.; McPhaul, M.J. Androgen receptor gene expression in human prostate carcinoma cell lines. Cancer Res. 1990, 50, 5382–5386.
- McEwan, I.J.; Brinkmann, A.O. Androgen Physiology: Receptor and Metabolic Disorders. In Endotext; Feingold, K.R., Ana-Walt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, H.J., Kaltsas, G., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000.
- Simental, J.A.; Sar, M.; Lane, M.V.; French, F.S.; Wilson, E.M. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J. Biol. Chem. 1991, 266, 510–518.
- Jenster, G.; van der Korput, H.; Trapman, J.; Brinkmann, A.O. Identification of Two Transcription Activation Units in the N-terminal Domain of the Human Androgen Receptor. J. Biol. Chem. 1995, 270, 7341–7346.
- Callewaert, L.; van Tilborgh, N.; Claessens, F. Interplay between Two Hormone-Independent Activation Domains in the Androgen Receptor. Cancer Res. 2006, 66, 543–553.
- Dehm, S.M.; Regan, K.M.; Schmidt, L.J.; Tindall, D.J. Selective Role of an NH2-Terminal WxxLF Motif for Aberrant Androgen Receptor Activation in Androgen Depletion–Independent Prostate Cancer Cells. Cancer Res. 2007, 67, 10067–10077.
- Kumar, R.; McEwan, I.J. Allosteric Modulators of Steroid Hormone Receptors: Structural Dynamics and Gene Regulation. Endocr. Rev. 2012, 33, 271–299.
- McEwan, I.J. Intrinsic disorder in the androgen receptor: Identification, characterisation and drugability. Mol. BioSyst. 2011, 8, 82–90.
- Lavery, D.N.; McEwan, I.J. Structural Characterization of the Native NH2-Terminal Transactivation Domain of the Human Androgen Receptor: A Collapsed Disordered Conformation Underlies Structural Plasticity and Protein-Induced Folding. Biochemistry 2008, 47, 3360–3369.
- Reid, J.; Kelly, S.M.; Watt, K.; Price, N.C.; McEwan, I.J. Conformational Analysis of the Androgen Receptor Amino-terminal Domain Involved in Transactivation. J. Biol. Chem. 2002, 277, 20079–20086.
- Davies, P.; Watt, K.; Kelly, S.; Clark, C.; Price, N.C.; McEwan, I.J. Consequences of poly-glutamine repeat length for the conformation and folding of the androgen receptor amino-terminal domain. J. Mol. Endocrinol. 2008, 41, 301–314.
- Meyer, S.; Wang, Y.-H.; Pérez-Escrivà, P.; Kieffer, B. Backbone 1H, 15N, 13C NMR assignment of the 518–627 fragment of the androgen receptor encompassing N-terminal and DNA binding domains. Biomol. NMR Assign. 2016, 10, 175–178.
- Eftekharzadeh, B.; Piai, A.; Chiesa, G.; Mungianu, D.; Garcia, J.; Pierattelli, R.; Felli, I.C.; Salvatella, X. Sequence Context Influences the Structure and Aggregation Behavior of a PolyQ Tract. Biophys. J. 2016, 110, 2361–2366.
- Li, J.; White, J.T.; Saavedra, H.; Wrabl, J.O.; Motlagh, H.N.; Liu, K.; Sowers, J.L.; Schroer, T.A.; Thompson, E.; Hilser, V.J. Genetically tunable frustration controls allostery in an intrinsically disordered transcription factor. eLife 2017, 6, e30688.
- Brodie, J.; McEwan, I.J. Intra-domain communication between the N-terminal and DNA-binding domains of the androgen receptor: Modulation of androgen response element DNA binding. J. Mol. Endocrinol. 2005, 34, 603–615.
- Dubbink, H.; Erik, J.; Hersmus, R.; Verma, C.; van der Korput, H.; Berrevoets, C.; van Tol, J.; der Made, A.Z.-V.; Brinkmann, A.; Pike, A.; et al. Distinct Recognition Modes of FXXLF and LXXLL Motifs by the Androgen Receptor. Mol. Endocrinol. 2004, 18, 2132–2150.
- Shaffer, P.L.; Jivan, A.; Dollins, D.E.; Claessens, F.; Gewirth, D.T. Structural basis of androgen receptor binding to selective androgen response elements. Proc. Natl. Acad. Sci. USA 2004, 101, 4758–4763.
- Haelens, A.; Tanner, T.; Denayer, S.; Callewaert, L.; Claessens, F. The Hinge Region Regulates DNA Binding, Nuclear Trans-location, and Transactivation of the Androgen Receptor. Cancer Res. 2007, 67, 4514–4523.
- Sahu, B.; Pihlajamaa, P.; Dubois, V.; Kerkhofs, S.; Claessens, F.; Jänne, O.A. Androgen receptor uses relaxed response element stringency for selective chromatin binding and transcriptional regulation in vivo. Nucleic Acids Res. 2014, 42, 4230–4240.
- Kerkhofs, S.; Dubois, V.; de Gendt, K.; Helsen, C.; Clinckemalie, L.; Spans, L.; Schuit, F.; Boonen, S.; Vanderschueren, D.; Saun ders, P.T.; et al. A role for selective androgen response elements in the development of the epididymis and the androgen control of the 5α reductase II gene. FASEB J. 2012, 26, 4360–4372.
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid Receptor Confers Resistance to Antiandrogens by Bypassing Androgen Receptor Blockade. Cell 2013, 155, 1309–1322.
- Kregel, S.; Bagamasbad, P.D.; He, S.; la Pensee, E.; Raji, Y.; Brogley, M.; Chinnaiyan, A.; Cieslik, M.; Robins, D.M. Differential modulation of the androgen receptor for prostate cancer therapy depends on the DNA response element. Nucleic Acids Res. 2020, 48, 4741–4755.
- Matias, P.M.; Donner, P.; Coelho, R.; Thomaz, M.; Peixoto, C.; Macedo, S.; Otto, N.; Joschko, S.; Scholz, P.; Wegg, A.; et al. Structural Evidence for Ligand Specificity in the Binding Domain of the Human Androgen Receptor. J. Biol. Chem. 2000, 275, 26164–26171.
- De Jésus-Tran, K.P.; Côté, P.-L.; Cantin, L.; Blanchet, J.; Labrie, F.; Breton, R. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci. 2006, 15, 987–999.
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 2012, 33, 887–894.
- Fuentes-Prior, P.; Rojas, A.; Hagler, A.T.; Estebanez-Perpina, E. Diversity of Quaternary Structures Regulates Nuclear Receptor Activities. Trends Biochem. Sci. 2019, 44, 2–6.


