 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lenie Van Rossem | + 2986 word(s) | 2986 | 2021-02-20 09:24:29 | | | |
| 2 | Peter Tang | Meta information modification | 2986 | 2021-03-07 09:40:35 | | |
Video Upload Options
Preconception folic acid supplement use is a well-known method of primary prevention of neural tube defects (NTDs). Obese women are at a higher risk for having a child with a NTD.
1. Introduction
In order to prevent neural tube defects (NTDs) in offsprings, women are advised to take a 0.4 mg folic acid supplement from the moment they wish to get pregnant up until the first trimester of pregnancy [1]. This advice applies to all women, except for women with a history of a previous child with a NTD, who are advised to take a higher dose of 4–5 mg folic acid supplement [1].
A growing number of women is obese when trying to get pregnant, with an increased risk of having a child with a NTD [2][3]. Meta-analyses showed a dose-response association between maternal Body Mass Index (BMI) and NTDs, and the risk rapidly increased in women with a BMI ≥ 30 kg/m2 (Table 1) [4][5][6]. In addition, a BMI ≥ 30 kg/m2, defined as maternal obesity, is also associated with the severity of the NTD in the offspring [7][8].
Table 1. Overview of three meta-analyses on the association between maternal obesity and NTD in offsprings.
| Years Included | Number of Studies | Design | Results (OR (95% CI)) | ||||
|---|---|---|---|---|---|---|---|
| Normal Weight | Overweight | Obese | Severely Obese | ||||
|
Rasmussen et al. 2008 [4] |
January 2000–January 2007 |
12 |
Cohort and case-control studies |
1 (ref) |
1.22 (0.99–1.49) |
1.70 (1.34–2.15) |
3.11 (1.75–5.46) |
|
Stothard et al. 2009 [5] |
January 1966–May 2008 |
18 |
Cohort and case-control studies |
1 (ref) |
1.87 (1.62–2.15) |
||
|
Huang et al. 2017[6] |
up to 15 December 2015 |
22 |
Case-control studies |
1 (ref) |
1.20 (1.04–1.38) |
1.68 (1.51–1.87) |
|
Given the known association between inadequate maternal folate intake and NTD in offsprings, and the increased risk of NTDs in obese women, the question arises whether obese women more often have a folate deficiency [9]. There might be an absolute folate deficiency from diet (folate) due to a suboptimal intake that is associated with obesity, combined with the fact that obese women may be less compliant in taking supplements (folic acid) [10][11][12]. On the other hand, obese women can have a relative folate deficient status, caused by a state of chronic low-grade inflammation, which results in an increased metabolic need of folate. Importantly, studies have shown that obese women had an increased risk of NTDs, regardless of their folate intake [13][14]. There are no studies that have assessed whether a high dose of folic acid results in less NTD pregnancies in obese women. Therefore, the rationale to prescribe higher doses of folic acid supplementation has to come from indirect evidence. Several underlying mechanisms have been suggested as determinants in the causal pathway of a relative folate deficiency in obese women, such as chronic inflammation and hyperinsulinemia [15]. However, an overview of causes of folate deficiency in obese women, potential underlying (patho)physiological mechanisms and how they might contribute to a higher risk of NTDs is lacking.
Moreover, different international recommendations on folic acid supplement use for obese women before and during pregnancy are used [16][17].
2. Epidemiology of Folate Deficiency in Obese (pre)Pregnant Women
2.1. Absolute Deficiency
Studies have shown that women with obesity have a lower intake of folate (Table 2). Women with obesity are less likely to use preconceptional folic acid supplement compared to normal weight women, 45.2% versus 60.4%, respectively [12]. They are also less likely to use folic acid supplements on a daily base, 26% versus 33%, respectively [10]. Moreover, women with obesity are less likely to receive enough folate through their diet than lean individuals, i.e., relative malnutrition [18][19]. Both a lower intake of folic acid supplements and a lower dietary intake of folate accounts for lower folate levels in serum, red blood cells, and body fluids. Moreover, decreased folic acid intake is often due to unplanned pregnancies and failed contraceptive methods prevalent in obese women [10].
Table 2. Intake of folate and folic acid supplements in women, per weight category.
| Study Design | Population | Sample Size | Outcome | Results (% or Mean ± SD) | ||||
|---|---|---|---|---|---|---|---|---|
| Normal Weight | Overweight | Obese | p-Value | |||||
|
Masho et al. 2016 [10] |
Cohort study |
Women with singleton pregnancy living in USA |
104.211 |
Daily intake of folic acid supplement |
33% |
29% |
26% |
<0.0001 |
|
Farah et al. 2013 [12] |
Cohort study |
White European women with a singleton pregnancy |
288 |
Use of folic acid supplement |
60% |
60% |
45% |
0.029 |
|
Bird et al. 2015 [18] |
Cohort study |
Non-pregnant women aged ≥19 years living in the USA |
538 |
Folate intake through diet (μg/L) |
559 ± 12.7 |
557 ± 14.5 |
517 ± 10.5 |
0.002 |
Though it is clear that obese women have a lower intake of folate, obesity is associated with other factors that are subsequently determinants of a lower intake of folate. Earlier studies indicated that smoking, lifestyle, age, parity, educational level, income level, and whether the pregnancy was planned were determinants of folate intake [20][21]. In a multivariable model, maternal weight status was independently associated with adequate use of folic acid, even after excluding women with an unplanned pregnancy [20].
2.2. Relative Deficiency
Obese women had lower serum folate levels even after controlling for folate intake through supplements and diet (β = −0.26, 95% CI: −0.54, 0.02); p = 0.07) [22]. When comparing non-obese and obese women with a similar folate intake, serum levels in obese women tend to be lower than in non-obese women, suggesting the current recommendations of folic acid supplement use could be subjected to review.
An increased need for folate is suggested to be caused by altered metabolic processes and chronic low-grade inflammation that could eventually underlie the increased risk for women with obesity on NTDs. Moreover, in women of higher weight categories, an adequate intake of folic acid of 0.4 mg/day did not lower the risk of NTDs [13]. A similar finding was reported by Parker et al., where women with obesity were at increased risk of NTDs, irrespective of adequacy of folic acid intake following the current standard ‘one-fits-all’ dosing regimens [14].
3. Pathophysiology of Relative Deficiency of Folate in Obese Women
3.1. Impaired One-Carbon Metabolism
Hyperhomocysteinemia, conventionally described as a serum level above 15 micromol/L, is a sensitive marker of an impaired one-carbon metabolism [23]. Considering the pathways within the one-carbon metabolism, a folate deficiency and as such less supply of methyl groups, contributes to higher levels of homocysteine, and higher levels of homocysteine lead to a higher demand for folate used for remethylation of homocysteine [24]. Moreover, hyperhomocysteinemia is a risk factor for several poor health outcomes, including, among others, neurological disorders, vascular diseases and reproductive disorders [25][26][27]. Pregnancy complications such as preeclampsia, intra-uterine growth restriction, and prematurity are associated with high maternal levels of homocysteine [25][28][29]. Hyperhomocysteinemia is more common in women with obesity, compared to non-obese individuals: two studies reported statistically significant differences in homocysteine levels between obese and non-obese women; 12.76 ± 5.30 μM/L versus 10.67 ± 2.50 μM/L, respectively, and 10.2 μM/L [4.6–26.3] versus 8.9 [4.4–25.8] respectively [30][31]. Suggested folate-related pathways that could underlie this finding are discussed below. In addition, an overview of potential underlying (patho)physiological pathways of folate deficiency and NTDs in obese women is displayed in Figure 1.
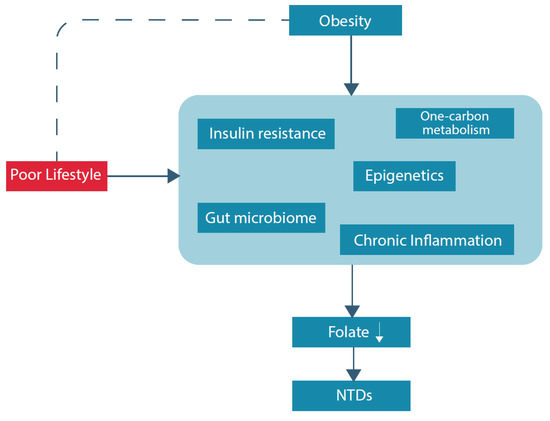
Figure 1. Overview potential underlying (patho)physiological pathways of folate deficiency and NTDs in obese women.
3.2. Physiology of Adipocytes
Adipose tissue is traditionally categorized into white and brown adipose tissue. Brown adipose tissue is specialized in energy expenditure and thermogenesis [32][33]. White adipose tissue is responsible for storing and releasing energy in the human body by controlling lipogenesis and lipolysis, respectively. During the process of lipogenesis, free fatty acids and glycerol are taken up from the blood stream and are stored as triglycerides in adipocytes [34]. On the contrary, lipolysis is the mechanism by which triglycerides are catabolized into free fatty acids and glycerol that are released into the bloodstream where they act as an energy source for other organs [35].
Obesity is characterized as an excessive growth of adipose tissue [36]. Furthermore, obesity is known to cause both hypertrophy as hyperplasia of the adipocyte [37]. These processes are associated with an infiltration of macrophages into the adipose tissue. This promotes inflammation and introduces TNFα into the tissue [38]. Moreover, the expansion of adipose tissue in obesity is linked to an inappropriate supply with oxygen and hypoxia development [39]. Subsequent inflammatory reactions inhibit preadipocyte differentiation and initiate adipose tissue fibrosis [40]. Not all obese individuals develop adipose tissue fibrosis followed by inflammation; however, obesity-related hypertrophic adipocytes may induce inflammation by producing pro-inflammatory adipokines [41].
3.3. Pro-Inflammatory State
The obesity-related low-grade chronic inflammation is generated by the production of pro-inflammatory cytokines, as IL-6 and TNF-α, and adipokines, as leptin [42]. Consumption of excess energy may as well acutely induce inflammatory responses [43][44]. Hence, it is thought that excess energy by overfeeding is another starting signal of inflammation, causing overactivation of tissues involved in metabolism, like adipose tissue, liver, and muscle, which in reaction to this stimulus provokes the inflammatory response [45][46]. Thus, besides continuous, low-grade chronic inflammation, there also might be additional, acutely induced inflammatory responses caused by excess supply of food. The inflammation-related collateral tissue damage activates tissue repair responses, requiring one-carbon moieties for synthesis of adequate amounts of proteins, lipids, nucleotides, and others. Since the folate dependent one-carbon metabolism supports cell proliferation at the detriment of B-vitamins, obesity-induced inflammation is associated with hyperhomocysteinemia and, thereby, folate deficiency. In addition, hyperhomocysteinemia is not only a result of inflammation, but hyperhomocysteinemia will again promote inflammation due to the excessive oxidative stress generated from high homocysteine levels [47].
3.4. Insulin Resistance
Adipose tissue regulates energy storage and release by lipogenesis and lipolysis. Obesity is associated with an increased basal lipolysis, which might be caused by an impaired sensitivity of adipocytes to insulin signaling, overexpression of the leptin gene in adipocytes, and increased circulating levels of leptin [48]. By the increased rate of lipolysis, higher amounts of fatty acids and glycerol are catabolized and enter the bloodstream. Increased serum levels of fatty acids, non-esterified fatty acids (NEFAs) in particular, are considered to be the most critical factor in inducing insulin resistance [35]. This is a pathological condition in which the capacity of cells to respond to normal levels of insulin is reduced. Increased NEFA levels are observed in persons with obesity and are associated with insulin resistance. Moreover, insulin resistance establishes within hours after an acute increase in plasma NEFA levels [49]. Beside the lipolysis-derived factors, the increased release of inflammatory cytokines influences the development of insulin resistance as well [50][51]. Especially, TNF-α and IL-6 cause an upregulation of potential mediators of inflammation that contribute to insulin resistance.
Additionally, chronic inflammation in general is not only associated with hyperhomocysteinemia and folate deficiency, but also with insulin resistance [52]. Although the exact working mechanism is not unravelled yet, it is suggested that insulin resistance influences activity of key enzymes in the folate dependent one-carbon metabolism, including 5,10-methylenetetrahydrofolate reductase (MTHFR) and cystathione b-synthase (CBS) [53][54]. Furthermore, it has been demonstrated that insulin signaling is affected by high levels of homocysteine, which is a condition associated with obesity [55][56]. Insulin signaling is an essential process in glucose homeostasis, since it increases the uptake of glucose into muscle and fat cells and reduces the synthesis of glucose in the liver. GLUT4 is one of the most important insulin-regulated glucose transporters responsible for decreasing blood glucose concentrations by facilitating glucose uptake into muscle and adipose tissue [57]. In the absence of insulin, the majority of GLUT4 is sequestered in intracellular vesicles in muscle and fat cells. When insulin levels increase, translocation of GLUT4 to the plasma membrane is induced and diffusion of circulating glucose down its concentration gradient into muscle and fat cells is facilitated. Homocysteine is one of the factors known to disrupt insulin signaling by impeding the GLUT4 translocation or recruitment on the plasma membrane and therefore reducing glucose uptake, which results in higher levels of glucose in the blood plasma [52].
3.5. Hyperglycaemia
Insulin resistance forces the pancreatic β-cells to produce more insulin to be able to prevent hyperglycaemia. However, when the compensatory insulin production is no longer sufficient, excessive amounts of glucose circulate in the blood plasma. This condition is referred to as hyperglycaemia, which is a defining characteristic of diabetes mellitus [58]. Besides maternal obesity, diabetes mellitus is a known risk factor for NTDs. Both obesity and diabetes mellitus are features of the metabolic syndrome [15]. The metabolic syndrome is further characterized by other metabolic risk factors including dyslipidemia, chronic hypertension, proinflammatory state, and prothrombotic state [59]. In the presence of 1 or 2 features of the metabolic syndrome, the fetus is on a 2-fold and 6-fold higher risk for NTD, respectively [60]. While the increased risk of NTDs associated with obesity appears to be independent of diabetes, a possible mechanism might be hyperglycemia due to insulin resistance in obese women [15].
Glucose levels are monitored and regulated by the islets of Langerhans in the pancreas and glucose is an essential factor for aerobic metabolism. Evidence suggests that the early developing embryo is dependent on maternal glucose metabolism, with detrimental effects in case of disbalance and hyperglycemia [61]. Thus, at the time of neural tube closure (around the fourth week of gestation), mothers with poorly regulated glucose levels are likely to have an suboptimal in utero environment, causing abnormal organogenesis [28][62][63]. To date, the exact working mechanism has not been elucidated yet. Only a few studies have reported evidence for this explanation, mostly focusing on the genetic susceptibility related to hyperglycemia as a risk factor for NTDs. Previous animal studies investigating molecular causes of NTDs in the embryos of diabetic mothers, demonstrated that in mouse embryos, expression of Pax3 is suppressed beginning on embryonic day 8.5 and subsequently, neuroepithelial cells undergo apoptosis and NTDs occur at increased frequency compared to embryos from nondiabetic pregnancies [64]. Moreover, in an embryos mouse model, which demonstrates a homozygous loss of function mutation in the Pax3 gene, NTDs can be rescued by either folic acid or thymidine supplementation [65][66]. This finding suggests that folic acid prevents NTDs by ensuring sufficient biosynthesis of factors for cell proliferation. Furthermore, a recent review of randomized controlled trials indicated that folic acid supplementation in non-pregnant populations, including women and men, had potential benefits on insulin resistance and glycemic control [67]. The mechanisms by which folic acid supplements lowers glucose levels and insulin resistance are still unclear. One of the suggested explanations is that hyperhomocysteinemia increases vascular oxidative stress, which could relate to insulin resistance and impaired insulin secretion during hyperglycemia [68][69]. As such, folate or folic acid supplements might decrease oxidative stress and, thereby, could prevent hyperglycemia and its detrimental effects.
3.6. Inositol
Inositol has been the focus of a large number of studies and is also involved in both folate uptake and glucose metabolism. Myo-inositol and D-chiro inositol are inositol isomers. Myo-inositol is the predominant form, which can be produced by the human body from D-glucose and is naturally present in foods, such as cereals, legumes, and meat [70]. Both isomeric forms of inositol were found to have insulin-like properties, acting as second messengers in the insulin intracellular pathway. Furthermore, both of these molecules are involved in increasing insulin sensitivity of different tissues, and thereby, improving health outcomes associated with insulin resistant, such as diabetes mellitus and reproductive disorders [71][72][73]. A randomized controlled trial showed that myo-inositol supplementation, started in the first trimester, in obese pregnant women reduced the incidence of gestational diabetes mellitus in the myo-inositol group compared with the control group, 14.0% compared with 33.6%, respectively (p = 0.001; odds ratio 0.34, 95% confidence interval 0.17–0.68) [74]. This reduction was achieved by improving insulin sensitivity.
Besides the insulin-like properties, an animal study demonstrated that myo-inositol is capable of significantly reducing the incidence of spinal NTDs in curly tail mice, a genetic model of folate-resistant NTDs [75]. Furthermore, in humans, significantly lower inositol concentrations have been reported in the blood of mothers carrying NTD fetuses compared with normal pregnancies, and mothers with low blood levels of inositol showed a 2.6-fold increased risk of an affected offspring [76].
Moreover, inositol is suggested to have preventive effects on NTD occurrence in curly tail mutant mouse [75]. Protection against diabetes-induced NTDs has been observed as well in other rodent models [77]. Hence, the animal data support a distinct inositol-dependent metabolic pathway that, when stimulated, can prevent NTDs.
3.7. Role of the Gut Microbiome
The gut microbiome can directly influence the folate status and via the cofactors vitamin B12 en B2, which contribute to a relative folate deficiency. The gut microbiome is the entirety of microorganisms, bacteria, viruses, protozoa, and fungi, and their collective genetic material present in the gastrointestinal tract [78]. For this overview, we focus on the bacterial microbiome. Gut bacterial microbiota are involved in a variety of essential processes, including the fermentation of indigestible food components into absorbable metabolites, the synthesis of essential vitamins, such as folate and vitamin B12, the removal of toxic compounds, the strengthening of the intestinal barrier, and the stimulation and regulation of the immune system [79][80][81]. Diversity is of great importance to a healthy intestinal microbiome, since it ensures redundancy, with multiple microbes competent to perform similar functions [82]. An imbalance in microbial populations, called dysbiosis, is associated with several poor health outcomes, including, among others, inflammatory bowel disease, neurological diseases, and diabetes [83][84]. Moreover, there is increasing evidence, mainly from animal studies, that alterations in the intestinal microbiome lead to metabolic and weight changes in the host [85][86].
An animal study found in genetically obese mice a 50% reduction in the abundance of Bacteroidetes and a proportional increase in Firmicutes [87]. Moreover, it is noted that changes affect the metabolic potential of the mouse gut microbiota. Previous research indicated that the obese microbiome has an increased capacity to harvest energy from the diet [86]. Furthermore, this trait is transmissible: colonization of germ-free mice with an ‘obese microbiota’ results in a significantly greater increase in total body fat than colonization with a ‘lean microbiota’. Besides the role of the gut microbiota as a contributing factor to the pathophysiology of obesity, it is also recognized as a source of B vitamins, in particular of folate and vitamin B12. It is produced by the colonic microbiota, mainly as the monoglutamate form of folate, the form that is absorbed at the highest rate. Thus, intestinal bacteria are a source of folate [88]. Even though absorption of folate occurs primarily in the duodenum and upper jejunum, the colon represents another depot of folate potentially affecting the general folate status of the host.
Moreover, the composition of the intestinal microbiome contributes to the regulation of intestinal permeability [89]. Short-chain fatty acids have been suggested as a mediator via which intestinal microbiota might promote the integrity of the intestinal mucosa. A higher intestinal permeability has been associated with obesity, leading to a ‘leaky gut’ with suboptimal uptake of micronutrients [90]. Hypothetically, there might be a derangement in the absorption of folate as well.
References
- World Health Organization. Standards for Maternal and Neonatal Care; World Health Organization: Geneva, Switzerland, 2007; p. 4.
- Poston, L.; Caleyachetty, R.; Cnattingius, S.; Corvalan, C.; Uauy, R.; Herring, S.; Gillman, M.W. Preconceptional and maternal obesity: Epidemiology and health consequences. Lancet Diabetes Endocrinol. 2016, 4, 1025–1036.
- Catalano, P.M.; Shankar, K. Obesity and pregnancy: Mechanisms of short term and long term adverse consequences for mother and child. BMJ 2017, 356, j1.
- Rasmussen, S.A.; Chu, S.Y.; Kim, S.Y.; Schmid, C.H.; Lau, J. Maternal obesity and risk of neural tube defects: A metaanalysis. Am. J. Obstet. Gynecol. 2008, 198, 611–619.
- Stothard, K.J.; Tennant, P.W.; Bell, R.; Rankin, J. Maternal overweight and obesity and the risk of congenital anomalies: A systematic review and meta-analysis. JAMA 2009, 301, 636–650.
- Huang, H.Y.; Chen, H.L.; Feng, L.P. Maternal obesity and the risk of neural tube defects in offspring: A meta-analysis. Obes. Res. Clin. Pr. 2017, 11, 188–197.
- Pace, N.D.; Siega-Riz, A.M.; Olshan, A.F.; Chescheir, N.C.; Cole, S.R.; Desrosiers, T.A.; Tinker, S.C.; Hoyt, A.T.; Canfield, M.A.; Carmichael, S.L.; et al. Survival of infants with spina bifida and the role of maternal prepregnancy body mass index. Birth Defects Res. 2019, 111, 1205–1216.
- Jensen, M.D.; Ryan, D.H.; Apovian, C.M.; Ard, J.D.; Comuzzie, A.G.; Donato, K.A.; Hu, F.B.; Hubbard, V.S.; Jakicic, J.M.; Kushner, R.F. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation 2014, 129, S139–S140.
- De-Regil, L.M.; Fernandez-Gaxiola, A.C.; Dowswell, T.; Pena-Rosas, J.P. Effects and safety of periconceptional folate supplementation for preventing birth defects. Cochrane Database Syst. Rev. 2010, 12, CD007950.
- Masho, S.W.; Bassyouni, A.; Cha, S. Pre-pregnancy obesity and non-adherence to multivitamin use: Findings from the National Pregnancy Risk Assessment Monitoring System (2009–2011). BMC Pregnancy Childbirth 2016, 16, 210.
- Hruby, A.; Manson, J.E.; Qi, L.; Malik, V.S.; Rimm, E.B.; Sun, Q.; Willett, W.C.; Hu, F.B. Determinants and Consequences of Obesity. Am. J. Public Health 2016, 106, 1656–1662.
- Farah, N.; Kennedy, C.; Turner, C.; O’Dwyer, V.; Kennelly, M.M.; Turner, M.J. Maternal obesity and pre-pregnancy folic acid supplementation. Obes. Facts 2013, 6, 211–215.
- Werler, M.M.; Louik, C.; Shapiro, S.; Mitchell, A.A. Prepregnant weight in relation to risk of neural tube defects. JAMA 1996, 275, 1089–1092.
- Parker, S.E.; Yazdy, M.M.; Tinker, S.C.; Mitchell, A.A.; Werler, M.M. The impact of folic acid intake on the association among diabetes mellitus, obesity, and spina bifida. Am. J. Obstet. Gynecol. 2013, 209, 239.e231–239.e8.
- Hendricks, K.A.; Nuno, O.M.; Suarez, L.; Larsen, R. Effects of hyperinsulinemia and obesity on risk of neural tube defects among Mexican Americans. Epidemiology 2001, 12, 630–635.
- Denison, F.C.; Aedla, N.R.; Keag, O.; Hor, K.; Reynolds, R.M.; Milne, A.; Diamond, A.; Royal College of Obstetricians and Gynaecologists. Care of Women with Obesity in Pregnancy: Green-top Guideline No. 72. BJOG 2019, 126, e62–e106.
- Vitner, D.; Harris, K.; Maxwell, C.; Farine, D. Obesity in pregnancy: A comparison of four national guidelines. J. Matern. Fetal Neonatal Med. 2019, 32, 2580–2590.
- Bird, J.K.; Ronnenberg, A.G.; Choi, S.W.; Du, F.; Mason, J.B.; Liu, Z. Obesity is associated with increased red blood cell folate despite lower dietary intakes and serum concentrations. J. Nutr. 2015, 145, 79–86.
- Parisi, F.; Rousian, M.; Steegers-Theunissen, R.P.M.; Koning, A.H.J.; Willemsen, S.P.; de Vries, J.H.M.; Cetin, I.; Steegers, E.A.P. Early first trimester maternal ’high fish and olive oil and low meat’ dietary pattern is associated with accelerated human embryonic development. Eur. J. Clin. Nutr. 2018, 72, 1655–1662.
- Camier, A.; Kadawathagedara, M.; Lioret, S.; Bois, C.; Cheminat, M.; Dufourg, M.-N.; Charles, M.A.; de Lauzon-Guillain, B. Social Inequalities in Prenatal Folic Acid Supplementation: Results from the ELFE Cohort. Nutrients 2019, 11, 1108.
- Barchitta, M.; Maugeri, A.; Lio, R.M.S.; Favara, G.; La Mastra, C.; La Rosa, M.C.; Agodi, A. Dietary Folate Intake and Folic Acid Supplements among Pregnant Women from Southern Italy: Evidence from the “Mamma & Bambino” Cohort. Int. J. Environ. Res. Public Health 2020, 17, 638.
- Knight, B.A.; Shields, B.M.; Brook, A.; Hill, A.; Bhat, D.S.; Hattersley, A.T.; Yajnik, C.S. Lower Circulating B12 Is Associated with Higher Obesity and Insulin Resistance during Pregnancy in a Non-Diabetic White British Population. PLoS ONE 2015, 10, e0135268.
- Blom, H.J.; Smulders, Y. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab. Dis. 2011, 34, 75–81.
- Collaboration, H.L.T. Lowering blood homocysteine with folic acid based supplements: Meta-analysis of randomised trials. Homocysteine Lowering Trialists’ Collaboration. BMJ 1998, 316, 894–898.
- Steegers-Theunissen, R.P.; Twigt, J.; Pestinger, V.; Sinclair, K.D. The periconceptional period, reproduction and long-term health of offspring: The importance of one-carbon metabolism. Hum. Reprod. Update 2013, 19, 640–655.
- Diaz-Arrastia, R. Homocysteine and neurologic disease. Arch. Neurol. 2000, 57, 1422–1427.
- Clarke, R.; Daly, L.; Robinson, K.; Naughten, E.; Cahalane, S.; Fowler, B.; Graham, I. Hyperhomocysteinemia: An independent risk factor for vascular disease. N. Engl. J. Med. 1991, 324, 1149–1155.
- Takao, Y.; Akazawa, S.; Matsumoto, K.; Takino, H.; Akazawa, M.; Trocino, R.A.; Maeda, Y.; Okuno, S.; Kawasaki, E.; Uotani, S.; et al. Glucose transporter gene expression in rat conceptus during high glucose culture. Diabetologia 1993, 36, 696–706.
- Bergen, N.E.; Jaddoe, V.W.; Timmermans, S.; Hofman, A.; Lindemans, J.; Russcher, H.; Raat, H.; Steegers-Theunissen, R.P.; Steegers, E.A. Homocysteine and folate concentrations in early pregnancy and the risk of adverse pregnancy outcomes: The Generation R Study. BJOG 2012, 119, 739–751.
- Vaya, A.; Rivera, L.; Hernandez-Mijares, A.; de la Fuente, M.; Sola, E.; Romagnoli, M.; Alis, R.; Laiz, B. Homocysteine levels in morbidly obese patients: Its association with waist circumference and insulin resistance. Clin. Hemorheol. Microcirc. 2012, 52, 49–56.
- Marchesini, G.; Manini, R.; Bianchi, G.; Sassi, S.; Natale, S.; Chierici, S.; Visani, F.; Baraldi, L.; Forlani, G.; Melchionda, N. Homocysteine and psychological traits: A study in obesity. Nutrition 2002, 18, 403–407.
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517.
- Townsend, K.; Tseng, Y.H. Brown adipose tissue: Recent insights into development, metabolic function and therapeutic potential. Adipocyte 2012, 1, 13–24.
- Verboven, K.; Wouters, K.; Gaens, K.; Hansen, D.; Bijnen, M.; Wetzels, S.; Stehouwer, C.D.; Goossens, G.H.; Schalkwijk, C.G.; Blaak, E.E.; et al. Abdominal subcutaneous and visceral adipocyte size, lipolysis and inflammation relate to insulin resistance in male obese humans. Sci. Rep. 2018, 8, 4677.
- Duncan, R.E.; Ahmadian, M.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 2007, 27, 79–101.
- Salans, L.B.; Cushman, S.W.; Weismann, R.E. Studies of human adipose tissue. Adipose cell size and number in nonobese and obese patients. J. Clin. Investig. 1973, 52, 929–941.
- Jo, J.; Gavrilova, O.; Pack, S.; Jou, W.; Mullen, S.; Sumner, A.E.; Cushman, S.W.; Periwal, V. Hypertrophy and/or Hyperplasia: Dynamics of Adipose Tissue Growth. PLoS Comput. Biol. 2009, 5, e1000324.
- Gustafson, B.; Gogg, S.; Hedjazifar, S.; Jenndahl, L.; Hammarstedt, A.; Smith, U. Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E999–E1003.
- Hosogai, N.; Fukuhara, A.; Oshima, K.; Miyata, Y.; Tanaka, S.; Segawa, K.; Furukawa, S.; Tochino, Y.; Komuro, R.; Matsuda, M.; et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 2007, 56, 901–911.
- Buechler, C.; Krautbauer, S.; Eisinger, K. Adipose tissue fibrosis. World J Diabetes 2015, 6, 548–553.
- DeBari, M.K.; Abbott, R.D. Adipose Tissue Fibrosis: Mechanisms, Models, and Importance. Int. J. Mol. Sci. 2020, 21, 6030.
- Mancuso, P. The role of adipokines in chronic inflammation. Immunotargets Ther. 2016, 5, 47–56.
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117.
- Valdearcos, M.; Xu, A.W.; Koliwad, S.K. Hypothalamic inflammation in the control of metabolic function. Annu. Rev. Physiol. 2015, 77, 131–160.
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445.
- Emanuela, F.; Grazia, M.; Marco de, R.; Maria Paola, L.; Giorgio, F.; Marco, B. Inflammation as a Link between Obesity and Metabolic Syndrome. J. Nutr. Metab. 2012, 2012, 476380.
- Mariotto, S.; Suzuki, Y.; Persichini, T.; Colasanti, M.; Suzuki, H.; Cantoni, O. Cross-talk between NO and arachidonic acid in inflammation. Curr. Med. Chem. 2007, 14, 1940–1944.
- Reynisdottir, S.; Langin, D.; Carlstrom, K.; Holm, C.; Rossner, S.; Arner, P. Effects of weight reduction on the regulation of lipolysis in adipocytes of women with upper-body obesity. Clin. Sci. 1995, 89, 421–429.
- Roden, M.; Price, T.B.; Perseghin, G.; Petersen, K.F.; Rothman, D.L.; Cline, G.W.; Shulman, G.I. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Investig. 1996, 97, 2859–2865.
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119.
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282.
- Kumar, A.; Palfrey, H.A.; Pathak, R.; Kadowitz, P.J.; Gettys, T.W.; Murthy, S.N. The metabolism and significance of homocysteine in nutrition and health. Nutr. Metab. 2017, 14, 78.
- Giltay, E.J.; Hoogeveen, E.K.; Elbers, J.M.; Gooren, L.J.; Asscheman, H.; Stehouwer, C.D. Insulin resistance is associated with elevated plasma total homocysteine levels in healthy, non-obese subjects. Atherosclerosis 1998, 139, 197–198.
- Gallistl, S.; Sudi, K.; Mangge, H.; Erwa, W.; Borkenstein, M. Insulin is an independent correlate of plasma homocysteine levels in obese children and adolescents. Diabetes Care 2000, 23, 1348–1352.
- Sanchez-Margalet, V.; Valle, M.; Ruz, F.J.; Gascon, F.; Mateo, J.; Goberna, R. Elevated plasma total homocysteine levels in hyperinsulinemic obese subjects. J. Nutr. Biochem. 2002, 13, 75–79.
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336.
- Ijuin, T.; Takenawa, T. Regulation of insulin signaling and glucose transporter 4 (GLUT4) exocytosis by phosphatidylinositol 3,4,5-trisphosphate (PIP3) phosphatase, skeletal muscle, and kidney enriched inositol polyphosphate phosphatase (SKIP). J. Biol. Chem. 2012, 287, 6991–6999.
- Martyn, J.A.; Kaneki, M.; Yasuhara, S. Obesity-induced insulin resistance and hyperglycemia: Etiologic factors and molecular mechanisms. Anesthesiology 2008, 109, 137–148.
- Grundy, S.M.; Brewer, H.B., Jr.; Cleeman, J.I.; Smith, S.C., Jr.; Lenfant, C.; American Heart, A.; National Heart, L.; Blood, I. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 2004, 109, 433–438.
- Ray, J.G.; Thompson, M.D.; Vermeulen, M.J.; Meier, C.; Wyatt, P.R.; Wong, P.Y.; Summers, A.M.; Farrell, S.A.; Cole, D.E. Metabolic syndrome features and risk of neural tube defects. BMC Pregnancy Childbirth 2007, 7, 21.
- Jovanovic-Peterson, L.; Peterson, C.M. Abnormal metabolism and the risk for birth defects with emphasis on diabetes. Ann. N. Y. Acad. Sci. 1993, 678, 228–243.
- Trocino, R.A.; Akazawa, S.; Takino, H.; Takao, Y.; Matsumoto, K.; Maeda, Y.; Okuno, S.; Nagataki, S. Cellular-tissue localization and regulation of the GLUT-1 protein in both the embryo and the visceral yolk sac from normal and experimental diabetic rats during the early postimplantation period. Endocrinology 1994, 134, 869–878.
- Maeda, Y.; Akazawa, S.; Akazawa, M.; Takao, Y.; Trocino, R.A.; Takino, H.; Kawasaki, E.; Yokota, A.; Okuno, S.; Nagataki, S. Glucose transporter gene expression in rat conceptus during early organogenesis and exposure to insulin-induced hypoglycemic serum. Acta Diabetol. 1993, 30, 73–78.
- Phelan, S.A.; Ito, M.; Loeken, M.R. Neural tube defects in embryos of diabetic mice: Role of the Pax-3 gene and apoptosis. Diabetes 1997, 46, 1189–1197.
- Fleming, A.; Copp, A.J. Embryonic folate metabolism and mouse neural tube defects. Science 1998, 280, 2107–2109.
- Wlodarczyk, B.J.; Tang, L.S.; Triplett, A.; Aleman, F.; Finnell, R.H. Spontaneous neural tube defects in splotch mice supplemented with selected micronutrients. Toxicol. Appl. Pharmacol. 2006, 213, 55–63.
- Zhao, J.V.; Schooling, C.M.; Zhao, J.X. The effects of folate supplementation on glucose metabolism and risk of type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Ann. Epidemiol. 2018, 28, 249–257.
- Weiss, N.; Heydrick, S.J.; Postea, O.; Keller, C.; Keaney, J.F., Jr.; Loscalzo, J. Influence of hyperhomocysteinemia on the cellular redox state--impact on homocysteine-induced endothelial dysfunction. Clin. Chem. Lab. Med. 2003, 41, 1455–1461.
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 2003, 52, 1–8.
- Clements, R.S., Jr.; Darnell, B. Myo-inositol content of common foods: Development of a high-myo-inositol diet. Am. J. Clin. Nutr. 1980, 33, 1954–1967.
- Croze, M.L.; Soulage, C.O. Potential role and therapeutic interests of myo-inositol in metabolic diseases. Biochimie 2013, 95, 1811–1827.
- Larner, J. D-chiro-inositol--its functional role in insulin action and its deficit in insulin resistance. Int. J. Exp. Diabetes Res. 2002, 3, 47–60.
- Genazzani, A.D.; Lanzoni, C.; Ricchieri, F.; Jasonni, V.M. Myo-inositol administration positively affects hyperinsulinemia and hormonal parameters in overweight patients with polycystic ovary syndrome. Gynecol. Endocrinol. 2008, 24, 139–144.
- D’Anna, R.; Di Benedetto, A.; Scilipoti, A.; Santamaria, A.; Interdonato, M.L.; Petrella, E.; Neri, I.; Pintaudi, B.; Corrado, F.; Facchinetti, F. Myo-inositol Supplementation for Prevention of Gestational Diabetes in Obese Pregnant Women: A Randomized Controlled Trial. Obstet. Gynecol. 2015, 126, 310–315.
- Greene, N.D.; Copp, A.J. Inositol prevents folate-resistant neural tube defects in the mouse. Nat. Med. 1997, 3, 60–66.
- Groenen, P.M.; Peer, P.G.; Wevers, R.A.; Swinkels, D.W.; Franke, B.; Mariman, E.C.; Steegers-Theunissen, R.P. Maternal myo-inositol, glucose, and zinc status is associated with the risk of offspring with spina bifida. Am. J. Obstet. Gynecol. 2003, 189, 1713–1719.
- Reece, E.A.; Khandelwal, M.; Wu, Y.K.; Borenstein, M. Dietary intake of myo-inositol and neural tube defects in offspring of diabetic rats. Am. J. Obstet. Gynecol. 1997, 176, 536–539.
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179.
- Heintz-Buschart, A.; Wilmes, P. Human Gut Microbiome: Function Matters. Trends. Microbiol. 2018, 26, 563–574.
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24.
- Engevik, M.A.; Morra, C.N.; Roth, D.; Engevik, K.; Spinler, J.K.; Devaraj, S.; Crawford, S.E.; Estes, M.K.; Kalkum, M.; Versalovic, J. Microbial Metabolic Capacity for Intestinal Folate Production and Modulation of Host Folate Receptors. Front. Microbiol. 2019, 10, 2305.
- Singer-Englar, T.; Barlow, G.; Mathur, R. Obesity, diabetes, and the gut microbiome: An updated review. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 3–15.
- Blanton, L.V.; Charbonneau, M.R.; Salih, T.; Barratt, M.J.; Venkatesh, S.; Ilkaveya, O.; Subramanian, S.; Manary, M.J.; Trehan, I.; Jorgensen, J.M.; et al. Gut bacteria that prevent growth impairments transmitted by microbiota from malnourished children. Science 2016, 351.
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel Dis. 2016, 22, 1137–1150.
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723.
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031.
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075.
- Kim, T.H.; Yang, J.; Darling, P.B.; O’Connor, D.L. A large pool of available folate exists in the large intestine of human infants and piglets. J. Nutr. 2004, 134, 1389–1394.
- Cox, A.J.; West, N.P.; Cripps, A.W. Obesity, inflammation, and the gut microbiota. Lancet Diabetes Endocrinol. 2015, 3, 207–215.
- Nagpal, R.; Newman, T.M.; Wang, S.; Jain, S.; Lovato, J.F.; Yadav, H. Obesity-Linked Gut Microbiome Dysbiosis Associated with Derangements in Gut Permeability and Intestinal Cellular Homeostasis Independent of Diet. J. Diabetes Res. 2018, 2018, 3462092.



