 +1 credit
+1 credit
Video Upload Options
Therapeutic proteins, such as growth factors (GFs), have been used in tissue engineering (TE) approaches for their ability to provide signals to cells and orchestrate the formation of functional tissue. However, to be effective and minimize off-target effects, GFs should be delivered at the target site with temporal control. In addition, protein drugs are typically sensitive water soluble macromolecules with delicate structure. As such, hydrogels, containing large amounts of water, provide a compatible environment for the direct incorporation of proteins within the hydrogel network, while their release rate can be tuned by engineering the network chemistry and density. Being formed by transient crosslinks, afforded by non-covalent interactions, supramolecular hydrogels offer important advantages for protein delivery applications.
1. Introduction
Tissue engineering (TE) has emerged as a result of bioengineering breakthroughs in the early 1990s [1] aiming to repair malfunction tissues in the body caused by genetic mutations, congenital abnormalities, aging, disease or injury. TE approaches are focused on the combined use of 1) cells, as building workers to repair and produce new tissue, 2) scaffolds, to support and guide cells and 3) biomolecules, which are able to regulate the fate of cells. Typically, those biomolecules are bioactive proteins, like growth factors (GFs), that regulate cell proliferation, differentiation, migration and other cell behaviors during tissue development [2][3]. The use of bioactive proteins has been widely exploited in TE, not only because of their direct effects on cells but also due to the rapid development of biotechnology, in particular the production of recombinant proteins. Direct administration of bioactive proteins in the body is known to be poorly controlled and can lead to undesired effects. Besides, the half-life of protein drugs in serum is very short, often within hours, requiring repeated dosing to maintain sufficient concentrations to produce therapeutic effects. To improve the availability of bioactive proteins, elegant delivery systems have been designed for their controlled and sustained release.
Hydrogels have become popular materials in biomedical applications due to their generally accepted biocompatibility and wide range of properties, from soft to stiff, to stimuli-responsive and cell-instructive. Hydrogels own a three-dimensional structure rich in water and held by a network of hydrophilic polymers. This architecture resembles the native extracellular matrix (ECM) in tissues. As such, hydrogels have been also highly considered for TE applications where they can hold cells [4] and provide mechanical support [5]. In addition, the properties of hydrogels offer various possibilities for the controlled delivery of proteins: 1) The massive water content enables the easy encapsulation of water-soluble molecules such as proteins; 2) The cross-linked network and composition of the hydrogels can be tailored, allowing control over the mesh size and thus the possibility to govern the release of entrapped proteins, based on their size and affinity to the hydrogel components; 3) The hydrated network provides protection to entrapped proteins against proteolytic degradation and prolongs their bioactivity. Based on the crosslinking method, hydrogels can be classified into two main types: chemically (through covalent bonds) and physically (or supramolecular) crosslinked hydrogels. Supramolecular hydrogels are formed via non covalent interactions such as hydrogen bonding, hydrophobic effects, host–guest recognition, electrostatic interactions, metal-ligand interaction, π-π interactions and van der Waals forces (Figure 1). Generally, supramolecular hydrogels are formed under mild environmental conditions, which enables the direct addition of sensitive molecules, such as proteins, during hydrogel formation. The dynamic nature of non-covalent interactions in supramolecular hydrogels, allows their minimally invasive delivery by injection. In addition, the dense crosslinked network can prevent diffusion of proteolytic enzymes and is thus believed to protect bioactive therapeutics from premature degradation [6]. The reversible nature of noncovalent crosslinking also provides repeated release on demand, as they are able to disassemble and reassemble based on environmental stimuli [7]. Figure 2 highlights the properties and medical applications of supramolecular hydrogels. Compared to supramolecular hydrogels, most hydrogels crosslinked by non-dynamic covalent bonds are unable to undergo crosslinking again after breaking and recover the original properties and function. Covalent bonding will decrease the flexibility of the hydrogels, making them difficult to integrate with the dynamic environment of native tissues [8][9]. Hence, the unique properties of dynamic and reversible noncovalent interactions make supramolecular hydrogels an ideal protein delivery system for TE applications. Table 1 provides a general comparison between hydrogels crosslinked by permanent covalent bonds and by supramolecular forces regarding their properties with relevance for protein delivery in TE applications.
Table 1. Comparison between hydrogels with permanent covalent and reversible crosslinks in relation to criteria relevant for protein delivery.
|
Criterion |
Covalently (Permanent Bonds) Crosslinked Hydrogels |
Supramolecular Hydrogels |
|
Processability |
More complex; covalent bonds form during hydrogelation requiring additional reagents or inputs (e.g., light source). |
Simple; it occurs spontaneously upon mixing hydrogel components or when in contact with electrolytes present in body fluids or culture medium. |
|
Injectability |
Difficult, unless if polymerizable in situ (e.g., under UV light). |
Common in most hydrogels due to their shear thining properties. |
|
Self-healing ability
|
Absent, except if made of dynamic covalent bonds (e.g., disulfide). |
Observed in most systems. |
|
Protein loading |
Mainly physical entrapment. Creating affinity interactions typically requires additional chemical modification of the hydrogel components. |
Various possibilities (physical entrapment + affinity interactions) not requiring additional chemical modification. |
|
Protein compatibility |
Risk of protein deactivation during hydrogel formation if subjected to denaturing agents (e.g., catalysts, UV light). |
Mostly compatible, unless there are strong electrostatic interactions between the protein and the hydrogel. |
|
Stability |
Stable at physiological conditions. |
Less stable at physiological conditions (e.g., can be affected by differences in ionic strength). |
This mini review will focus on the use of supramolecular hydrogels for the delivery of proteins in TE. We start by describing the various types of supramolecular hydrogels and how they have been designed, even if their applications in TE have not been demonstrated. The aim is to provide an overview on the repertoire of supramolecular hydrogels reported in the literature and with potential utility in TE. The possibility to control the kinetics of protein release from supramolecular hydrogels are discussed through examples utilizing model proteins. Next, the application of supramolecular hydrogels for the controlled release of bioactive proteins in the context of TE is described including angiogenesis, bone and cartilage regeneration and wound healing. Finally, supramolecular hydrogel/protein formulations that have reached clinical trials are shortly discussed to provide a better understanding of the clinical application prospects of supramolecular hydrogels.
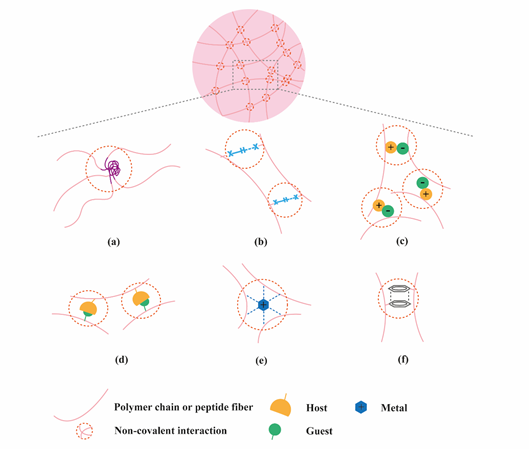
Figure 1. Application of supramolecular chemistry to create physically crosslinked hydrogels. (a) hydrophobic effects; (b) hydrogen bonding; (c) electrostatic interactions; (d) host-guest interactions; (e) metal ligand interactions; (f) π-π stacking.
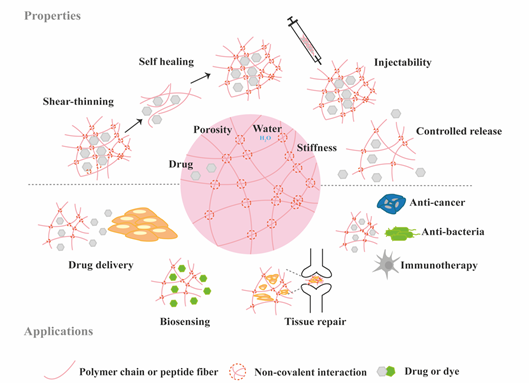
Figure 2. Schematic highlighting the properties and medical applications of supramolecular hydrogels.
2. Classification of Supramolecular Hydrogels Based on Their Composition
2.1. Polymer-Based Hydrogels
Polymer-based supramolecular hydrogels can be from natural or synthetic origin. The most popular advantages of natural polymers are their biocompatibility and biodegradation which are key in TE applications. Polysaccharides are a series of hydrophilic natural polymers including dextran, chitosan, hyaluronic acid, alginate, cellulose among others.
Dextran (Dex) is a water-soluble polysaccharide consisting of α-1,6-linked D-glucopyranoses and the hydroxyl groups in dextran can be conjugated with functional groups for the formation of a crosslinked structure. Chen et al. [10] conjugated dextran with 2-naphthylacetic (2-NAA) through ester bond and hyaluronic acid with β-cyclodextrin (β-CD) to form supramolecular hydrogel (HA-Dex) by host-guest interaction between β-CD 2-NAA. Cell culture experiments demonstrated that NIH-3T3 fibroblasts could adapt to the microenvironment formed by HA-Dex hydrogels making HA-Dex a potential material as cell scaffold. Dextran can also be modified into carboxymethyl dextran (CMDH) and subsequently to aminodextran (AD) which can then be utilized as additives with a derived C2-phenylalanine gelator (LPF) [11]. LPF interacted with CMDH and AD via hydrogen bonding and π-π stacking respectively, resulting in enhanced mechanical stability of the hydrogel.
Chitosan is linear polysaccharide with cationic nature, composed of randomly distributed β-(1-4)-linked d-glucosamine and N-acetyl-D-glucosamine units [12]. Free amino and hydroxyl groups of chitosan can be easily modified to incorporate functional groups amenable for supramolecular interactions. An injectable supramolecular carboxymethyl chitosan-zinc (CMCh-Zn) hydrogel was prepared for antibacterial applications [13]. The coordination of empty orbitals of Zn2+ with lone pair of electrons of NH2, OH and COO− groups of CMCh leads to the rapid formation of CMCh-Zn complex after simply mixing a solution of modified chitosan and Zn(NO3)2·6H2O salt together within the pH range of 5.3–7.0. These CMCh-Zn hydrogels could be used for bone TE applications as Zn is an essential element in bone homeostasis and has been used as a therapeutic agent in bone regeneration [14][15]. However, the use of metal ions should be carefully considered as they can be toxic if exceeding tolerable concentrations.
Hyaluronic acid (HA) is a linear polysaccharide composed of repeating disaccharide units of d-glucuronic acid and N-acetylglucosamine. Burdick`s group developed an HA-based supramolecular hydrogel based on β-CD-modified HA (HA-β-CD) and adamantine-modified HA (HA-Ad) through host-guest interactions between CD and Ad [16]. This HA-based hydrogel is shear-thinning and could rapidly recover its gel form at injection site, indicating its great potential for non-invasive delivery. The hydrogel was upgraded by modification of HA with azobenzene (Azo), a light sensitive molecule, instead of Ad [17]. The host-guest interaction between CD and Azo could be modulated by light with different wavelength, as shown in Figure 3a, which was able to tune the release of entrapped protein. HA was also modified with a hydrophobic molecule to obtain amphiphilic HA. Cholesterol was conjugated to HA as building block, which could self-assemble into an injectable nanohydrogel [18][19]. The self-assembly ability of cholesterol-grafted-HA (HA-CH) appears owing to the hydrophobic interactions between the cholesterol cores and the hydrophilic interactions between HA shells. In addition, due to the presence of hyaluronidase in tissues, HA-based hydrogels are able to be enzymatically degraded to achieve a complete release of the entrapped cargos [20].
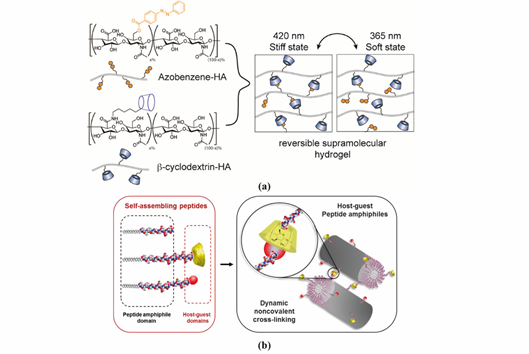
Figure 3. Representative supramolecular hydrogels based on host-guest interaction. (a) Polymer-based host-guest hydrogel between HA-β-CD and HA-Azo. With UV-irradiation at 365 nm, azobenzene will transform from its trans-state to cis-state, leading to gel disassembly. The gelling process is reversible with irradiation of visible light (400–500 nm). Adapted with permission from Ref. [17] Copyright © (2018), American Chemical Society. (b) Peptide-based host-guest hydrogel between PA-β-CD and PA-Ad. Incorporating β-CD and Ad interaction in the PA nanofiber improves the stability of PA-based hydrogel. Adapted with permission from [21] Copyright © (2019), American Chemical Society.
Synthetic polymers can be designed and synthesized with specific functional groups to obtain hydrogels with on-demand physical properties. The synthetic polymer poly(ethylene glycol) (PEG) has been widely investigated in TE applications due to its easy functionalization. A great number of supramolecular hydrogels were prepared via PEG-based polymers. β-CD and cholesterol were conjugated to star-shaped PEGs and supramolecular hydrogels were shown to self-assemble via hydrophobic and van der Waals interactions between β-CD and cholesterol [22]. The ability of the star-shaped PEG based hydrogels as protein delivery vehicles and the release profile of model proteins from these hydrogels were then investigated. The cross-link density and swelling stresses played prominent roles in controlling the release kinetic. On the one hand, after absorption of water, the increased swelling stresses accelerated the dissociation of β-CD/cholesterol complexes. On the other hand, the flexible polymer was able to relieve some swelling stresses to slow down the dissociation of the complexes. Hence, a nearly zero-order release of the entrapped proteins was achieved with the balance between the two mechanisms [23].
Another class of supramolecular hydrogels receiving great attention in drug delivery applications are based on polymer–CD inclusion complexes [24]. It has been shown that hydrophilic polymers such as PEG could penetrate the inner cavity of α-CD forming an inclusion complex with a necklace-like structure [25]. These polymer–CD inclusion complexes can self-assemble, via aggregation of the inclusion domains, and lead to the formation of a physically crosslinked hydrogel (Figure 4a). In these systems, drug incorporation can be achieved in aqueous environment during the gelation process making it attractive for protein delivery. Using poly(caprolactone)-poly(ethylene glycol)-poly(caprolactone) (PCL-PEG-PCL) triblock copolymer and γ-cyclodextrin (γ-CD), an injectable supramolecular hydrogel was developed for insulin delivery [26]. The inclusion complexes were formed directly by the PEG segment in PCL-PEG-PCL backbone and γ-CD, not requiring conjugation with additional guest molecules. The ratio between PEG and PCL determined the formation of the hydrogel. A certain amount of hydrophilic PEG could keep a balance between hydrophobic (PCL) and hydrophilic (PEG) segments of the copolymer and increase the chance of γ-CD to thread onto the PEG blocks, since hydrophobic interaction between PCL segments acts as a barrier against γ-CD threading. PEG blocks were covered by γ-CD when inclusion complexes were formed, thus enhancing the opportunity of hydrophobic interactions via PCL segments, leading to the rapid gel formation (Figure 4b).
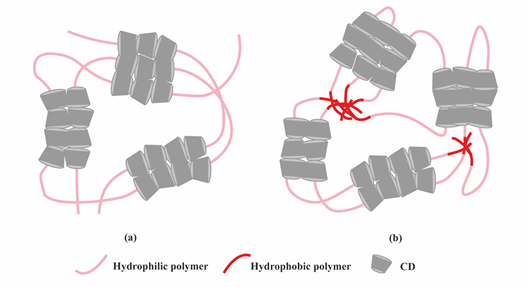
Figure 4. Scheme showing the formation of supramolecular hydrogels by polymer–CD inclusion complexes. (a) Threading of CD onto hydrophilic polymers; (b) Threading of CD onto amphiphilic polymers.
Stimuli-responsive hydrogels have been also exploited for the delivery of therapeutic proteins. These smart hydrogels undergo phase transition in response to external changes in the environment such as temperature, pH, light, magnetic [27] and electrical fields [28]. The external stimuli can be accurately regulated to achieve precise control over protein release.
Synthetic polymers with lower critical solution temperature (LCST) are suitable for design of injectable thermo-sensitive hydrogels, including poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide) (PEO-PPO-PEO) [29], poly(vinyl ether)s (PVEs) [30] and a series of N-substituted acrylamide polymers such as poly(N-isopropylacrylamide) (PNIPAm), poly(N,N-diethylacrylamide) (PDEAm), poly(N-vinyl-n-butyramide) (PNVBAm), among others. For example, PNIPAm has an LCST at 32 °C, which is higher than room temperature, but lower than body temperature, meaning PNIPAm can easily reach sol-gel transition after injection in the body. Modification of PNIPAm with acryloyl-β-cyclodextrin (Aβ-CD) was found to decrease the LCST to 28-30 °C with different conjugation rates, indicating LCST can be slightly affected by modification of the thermo-sensitive polymer [31]. β-CD modified PNIPAm and adamantyl-terminated poly(ethylene glycol) (Ad-PEG) were synthesized to form dual supramolecular assemblies using the host-guest interaction between β-CD and adamantyl group, together with the formation of polypseudorotaxane between α-cyclodextrin (α-CD) and PEG chains with additional α-CD added into the system [32]. When the temperature increased from 25 to 37 °C, the hydrophobic interactions of PNIPAm segments would become the dominant force, making hydrogels stronger. Conversely, a thermo-sensitive response could also contribute to a controlled release profile when the hydrogels undergo gel-sol transition. Another thermo-sensitive hydrogel using host–guest interaction was prepared using an amphiphilic copolymer pyrene-poly(caprolactone)-b-poly(oligo(ethylene glycol) methacrylate) (Py-PCL-b-POEGMA) and α-CD at room temperature [33]. α-CD acted as the host molecule while POEGMA acted as guest molecule. BSA was loaded in this thermo-sensitive hydrogel and a faster release at 37 °C was achieved compared to 25 °C. The temperature-dependent behavior of the release results from the dissociation of α-CD when the temperature is increased. Therefore, the hydrogels suffered partly from structural damage at higher temperature and faster release was observed.
Similar to temperature responsiveness, pH changes were also utilized to trigger phase transitions of supramolecular polymer hydrogels taking advantage of pH differences in different parts of body. Hydrogen bonds and electrostatic interactions are pH-sensitive. Changes in pH affect the protonation/deprotonation of acidic/basic groups on polymers. For example, a synthetic, catheter-injectable supramolecular hydrogel was fabricated by ureido-pyrimidinone (UPy) units and poly(ethylene glycol) (PEG) chains via hydrogen bonding. These UPy-modified PEG hydrogels formed fibers in aqueous solution and were able to undergo gel-sol transition at basic pH, and reversibly turn back to gel state at neutral pH. The fast pH switchability of UPy-modified PEG hydrogels was likely caused by the breaking down of crosslinks between the fibers that form the transient network instead of complete disassembly of the fibers [34]. A pH-responsive supramolecular hydrogel was prepared based on poly(acryloyl 6-aminocaproic acid) (PA6ACA) and poly(methacrylic acid-co-ethyl acrylate) (EUDRAGIT L 100-55). In the acidic environment, intermolecular hydrogen bonds between carboxyl groups and amide units on PA6ACA and EUDRAGIT L100-55 drove the assembly of the supramolecular network. At neutral aqueous environments, the hydrogen bonds were eliminated due to the deprotonation of carboxylic acid groups, leading to the gel-sol transition [35].
Another strategy to build stimuli triggered supramolecular hydrogels is to modify the polymer with functional groups that are sensitive to light. Light stimulus is different from temperature or pH stimuli since it can be precisely controlled outside the body. One example that has been widely exploited is the use of cis-trans isomerization to generate light-responsive supramolecular hydrogels. The phase transition of this kind of hydrogel is usually based on host-guest interaction. As previously mentioned, HA was functionalized with CD and Azo to obtain a light-responsive supramolecular hydrogel. Tamesue et al. [36] reported a similar light responsive supramolecular hydrogel prepared by CD-conjugated curdlan (CD-CUR) and Azo-modified poly(acrylic acid)(pAC12Azo). The supramolecular hydrogels were fabricated under visible light and underwent gel-sol transition upon irradiation at 365 nm, which resulted in the transition from trans isomer to cis isomer of Azo group. Recently, the CD- and Azo-based light-responsive supramolecular hydrogel was modified by using upconverting nanoparticles [37], in order to induce the phase transition of hydrogel under NIR irradiation, which has a deeper tissue penetration and is safer for in vivo applications than UV light. Derivatized Azo (modified with an alkoxypropanol (azopropOH) side group) was conjugated to poly(acrylic acid) which interacted with deoxycholate-β-CD to form host-guest complex. LiYF4:Tm3+/Yb3+ upconverting nanoparticles (LnUCNPs) were immobilized in the hydrogel matrix to provide upconversion effect in UV-region. Due to the high viscosity environment of the supramolecular hydrogel, the Brownian motion of LnUCNPs was limited which increased the chance for energy transfer to achieve phase transition. The unmodified supramolecular hydrogel went through the gel-sol transition under 365 nm irradiation in 25 min. When incorporated with LnUCNPs, the resulting light responsive supramolecular hydrogel was found to have a gel-sol transition time of 60 min under 980 nm excitation. Although a long time was needed, the successful NIR response indicates its potential to act as photo-responsive delivery system for applications in deep tissue. Another strategy to achieve light sensitivity is to use photo-cleavable moieties, but this strategy is often used to prepare chemically crosslinked hydrogels and will not be discussed in this review.
2.2. Peptide-Based Hydrogels
The self-assembly ability of peptides has attracted great attention to develop supramolecular hydrogels. Peptides have been engineered with various molecular motifs (α-helix, β-sheet, amphiphilic, collagen- and elastin-like) to self-assemble into filaments, which at certain concentration can entangle and form a nanofibrous hydrogel network.
A designed supramolecular hydrogel formed by hydrogelating self-assembling fibers (hSAFs) was reported by Mehrban et al. [38]. Two peptides gelled together and formed coiled-coil α-helical fibrous nanostructures. Subsequently, the cell adhesion motif RGDS was attached to the peptide fibers containing azide functionality via a click reaction with alkyne-RGDS for integrin binding. Images from scanning electron microscopy (SEM) showed interconnected fibers and porous structure in both hydrogels with or without RGDS, indicating the stability of coiled-coil fibrous structures. Similar approach could be used to attach protein molecules onto hSAFs.
Peptides designed to self-assemble with β-sheet structure normally requires repeat sequences of ionic hydrophilic and hydrophobic amino acids, such as AEAEAKAKAEAEAKAK (AEAK16-II) [39]. The peptide sequence forms β-sheet structure with hydrophobic face on one side and hydrophilic face on the other side, with the hydrophobic in the fiber core contributing to the stability of the structure. The electrostatic interactions and hydrogen bond between β-sheet layers results in the formation of fibrils. Both small molecules and biomacromolecules could be entrapped between these fibrils for sustained release by modulating the fiber density. A two-layered nanofiber hydrogel was formed by Ac-(RADA)4-NH2 and Ac-(KLDL)3-NH2 self-assembling peptides with Ac-(RADA)4-NH2 in the core layer and Ac-(KLDL)3-NH2 in the shell layer. The mechanical properties, as well as the hydrogel network density, could be altered by adjusting the density of Ac-(KLDL)3-NH2. In addition, the initial burst release of protein from this two-layer hydrogel was decreased compared to the single peptide formed hydrogel, which resulted from the higher nanofiber density provided by the additional layer [40]. The morphology of a self-assembled β-sheet pentapeptide hydrogels could be tuned by altering the charge distribution of the peptide sequence [41]. The pentapeptide contains three aliphatic isoleucine (I) residues, with potential to form β-sheets, and two aspartic acid (D) residues to improve solubility (DIIID-NH2, DDIII-NH2 and IDIDI-NH2). These three pentapeptide sequences can form robust hydrogels with gelation induced through changes in pH. Morphology examination by cryo-focused ion beam SEM showed IDIDI-NH2 hydrogels were formed by high aspect-ratio nanofibers while the DDIII-NH2 and DIIID-NH2 hydrogels were made of more entangled and interconnected structures, indicating that small alterations in the sequence can cause significant changes in the structure of resulting gels.
Peptide amphiphiles (PAs) are another class of self-assembling building blocks for hydrogel formation. PAs can be of three subclasses: 1) amphiphilic peptides; 2) lipidated peptides and 3) PAs conjugated with supramolecular binding motifs [42].
Amphiphilic peptides are composed of amino acids only. The balance between hydrophobic and hydrophilic forces largely contributes to the self-assembly process of amphiphilic peptides. A pH-responsive supramolecular peptide hydrogel was self-assembled from a synthetic peptide called PEP-1 (Ac-FALNLAKD-NH2) [43]. In the PEP-1 sequence, F, A and L amino acid residues are hydrophobic while D, N and K are hydrophilic, making PEP-1 an amphiphilic peptide. PEP-1 was able to form hydrogel at pH 7.4 due to the electrostatic interaction between aspartic acid (D) and lysine (K) residues, but the structure could be destroyed either in acidic or basic environments (pH 5.5, 9.0 and 12.0). In acidic environment, the protonation of the carboxylates in aspartic acid was not able to hold the electrostatic interaction with lysine amine groups and keep the entangled nanofibers, while in the basic environment, the increased solubility of PEP-1 and electrostatic repulsion between aspartic acid residues may be responsible for the lack of well-defined assembly.
Lipidated peptides are hybrid molecules consisting a hydrophobic alkyl (lipid) tail and a peptide segment containing, or not, sequences to form secondary structures, and a hydrophilic head to enhance water solubility. This class of PAs have been widely reported in the literature due to their design versatility and diversity of self-assembled nanostructures [44]. As such, they offer great potential to create a range of biomaterials for different biomedical applications, from drug delivery to TE [45]. Many PAs are designed to contain a β-sheet forming segment in order to promote their self-assembly into nanofiber structures. An injectable hydrogel was prepared based on palmitoyl-GNNQQNYKD-OH PA. Incorporation of the triptolide drug did not affect the hydrogel formation [46].
PA conjugates, consisting of PA molecules bearing supramolecular motifs at the C-terminus were recently reported to enable noncovalent cross-linking between PA nanofibers (Figure 3b). β-CD and Ad were coupled to a cationic PA (palmitoyl-V3A3K3), separated by a glycine spacer (G3), by copper(I)-catalyzed alkyne−azide cycloaddition. The resulting supramolecular hydrogel showed enhanced mechanical properties and resistance to degradation. Hydrogels formed by PA-DNA conjugate nanofibers cross-linked by DNA hybridization were also reported by the Stupp group [47]. Oligonucleotides were covalently linked to a lysine side chain at PA C-terminal by click chemistry to obtain PA-DNA conjugates, which was then co-assembled with a filler PA. Their co-assembly at different molar concentrations results into nanofibers displaying single-stranded DNA at different densities. Mixing fibers containing complementary DNA strands generates a reversible hydrogel which could disassemble when soluble single-stranded DNA is added as consequence of the toehold-mediated strand displacement mechanism. The dynamic organization of the nanofibers within the hydrogel network was shown to modulate phenotypic transformations in astrocytes.
Selection of supramolecular hydrogels using polymer or peptide building blocks requires some considerations from the development and the application point of view. We have attempted to identify advantages and disadvantages associated with both types of hydrogels (Table 2).
Table 2. Pros and Cons of polymer- and peptide-based hydrogels.
|
Type of Hydrogels |
Pros |
Cons |
|
Polymer-based |
• Great diversity of building blocks among synthetic and natural polymers • Tunable mechanical properties via synthetic polymer (e.g., molecular weight, copolymer design) • Good biostability • Easily modified through a variety of functional groups available (e.g., carboxylic, hydroxyl) • Easily controlled by stimuli |
• Potential toxicity of synthetic polymers • Lack of bioactivity or biodegradability of some polymers |
|
Peptide-based |
• Easily designed and synthesized • Easily modified through carboxylic or amino groups for the incorporation of other supramolecular moieties • Nanofibrous network formation resembles natural ECM structure • Biodegradable • Non-toxic • Some peptides have intrinsic bioactivity |
• Weak mechanical properties and not adequate for certain TE applications • pH related solubility • Less stable • Protein inactivation due to strong peptide-protein interactions |
2.3. Nucleic Acid-Based Hydrogels
Although not widely exploited as polymers and peptides, nucleic acids (mainly DNA) are gaining significant attention as building blocks for the supramolecular fabrication of hydrogels. Hydrogels can be formed by reversible cross-linking through DNA self-assembly (two complementary single-stranded DNA molecules can form a single double-stranded molecule through Watson-Crick base pairing, a process known as DNA hybridization) and can consist entirely of DNA or short DNA sequences grafted onto polymer backbones. Furthermore, using protein-binding aptamers, proteins can be captured within the DNA-based hydrogel (Table 1) and their release initiated using the displacement strand strategy. However, the use of DNA strands as the release trigger may not feasible in in vivo applications. Two recent reviews provide insightful background on the design, properties and biomedical applications of supramolecular DNA-based hydrogels [48][49] and thus this type of hydrogels will not be discussed in detail here.
The formation of hydrogels using nucleopeptides was reported the Xu and collaborators where nucleobases (thymine, adenine, cytosine, and guanine) were conjugated at the N-terminus of short peptides (FF, FFY, FFYp) [50]. The nucleopeptides were able to self-assemble in water, upon a pH- or enzyme trigger, and were shown to be resistant to proteinase K, a proteolytic enzyme. The self-assembled nucleopeptide hydrogels supported cell migration. Following a similar conjugation approach, the group of Laura Suggs screened a nucleo-tripeptide library for their ability to form hydrogels at physiological conditions [51]. The mechanical properties of the hydrogels varied from 10 Pa to 1 kPa depending on the nucleobase and amino acid composition. Oligonucleotides (length of 19 bases) have been also conjugated at the C-terminus of the self-assembling Fmoc-FF-OH peptide using copper-free click chemistry to yield pepDNA19 [52]. Mixing peptides bearing complementary oligonucleotides promoted nanofiber bundling which could lead to gel formation. PepDNA19 assemblies were sensitive to pH changes and could be degraded by DNase. More recently, the impact of C-terminus chemistry on the self-assembly of guanosine (gs)-containing nucleopeptides (gs-GKFF) was investigated [53]. The self-assembly was governed by the peptide segment, forming β-sheet structures, with the hydrogen-bonded guanosine (G-quartet or G-ribbon) contributing with additional secondary structures within the peptide conformation. The morphologies of the nucleopeptides assemblies were shown to depend on the C-terminus chemistry (amide or carboxylic acid).
Combining Nature’s building blocks in a single molecule, a nucleobase (thymine, cytosine, adenine, or guanine) linked to an amino acid (one or two phenylalanine) and glycoside (d-glucosamine) Xu’s group designed a new class of supramolecular hydrogelator [54], which were shown to self-assemble in water and form hydrogels at concentration at 3 wt%. The hydrogels exhibited viscoelastic properties, reaching storage modulus of 220 kPa, and stability in presence of proteolytic enzymes. They were able to bind and deliver nucleic acids to cells. By expanding the range of building blocks for fabricating supramolecular hydrogels, novel functional materials with new properties can be discovered for applications in TE.
2.4. Multi-Component Hydrogels
More recently, combining peptides with either natural or synthetic polymers has resulted in a new class of hybrid supramolecular hydrogels aiming to improve their mechanical properties and/or improve their biological or chemical responsiveness [55]. In general, hydrogels fabricated by peptides on their own present low mechanical properties [56]. At the same time, peptides are able to enhance the gelation process, preventing polymer aggregation [57], as well as providing a source of therapeutic molecules.
In this section, we defined “hybrid” as a multi-component supramolecular hydrogel formed via physical interaction between polymers and peptides or modified peptides, instead of self-assembly of hybrid lipopeptides, such as PAs. One of the earliest report on the formation of hybrid supramolecular hydrogels is the self-assembly of a heparin-binding PA with heparin reported by the Stupp group [58]. Gel formation was attributed to the electrostatic interaction between the negatively charged heparin chains and the positively charged PA with the sequence of C16-AAAAGGLRKKLGKA. The PA molecule presented an initial α-helix structure, but changed to β-sheet conformation after addition of heparin, which contributed to the PA self-assembly into nanofibers and gel formation.
Electrostatic interaction is the most popular driving force to form hybrid supramolecular hydrogels. N-fluorenylmethoxycarbonyl diphenylalanine (Fmoc-FF-OH) and poly-l-lysine (PLL) were gelled together to form an injectable supramolecular hydrogel. Fmoc-FF-OH is able to self-assemble into β-sheet structure but with poor rheological properties. When combined with PLL, the electrostatic interactions between positively charged PLL and negatively charged Fmoc-FF-OH nanofiber result in an enhancement of mechanical properties. Furthermore, thiol groups were introduced into PLL to improve the stability of hydrogels. Through various interactions, such as hydrophobic interaction, electrostatic forces, π-π stacking and hydrogen bonding, the PLL-SH/ Fmoc-FF-OH hydrogel could present nanofibers in helical conformation with amphipathic and amphoteric behavior. Borges et al. [59] reported a hybrid peptide/polymer supramolecular hydrogel combining self-assembly and layer-by-layer (LbL) assembly technique. Low molecular weight PA with sequence lauryl-VVAGKKK-NH2 (K3PA) was synthesized consisting of a hydrophobic lauryl tail, a hydrogen bonding sequence and a positively charged hydrophilic sequence. This PA was able to interact with anionic high molecular weight alginate (ALG) biopolymer via electrostatic interaction. Then, quartz crystal microbalance with dissipation monitoring technique was applied to study the nanofilm build-up process. A longer adsorption time was needed for the deposition of K3PA molecule compared with ALG, showing that the binding and arrangement of K3PA was slow. Although not reported in this study, multiple GFs could be loaded during multi-layer build-up for sequential GF co-delivery [60]. Using the negatively charged synthetic polymer poly(sodium 4-styrenesulfonate) (PSS) and a positively charged PA (palmitoyl-V3A3K3-NH2) a hybrid supramolecular hydrogel was reported recently (Figure 5) [61]. Upon mixing PA with PSS, self-supporting opaque hydrogels were formed within minutes. Rheology tests demonstrated the formation of stiff PSS/PA hydrogels and their stiffness and stability could be tuned by adjusting the chain length of PSS.
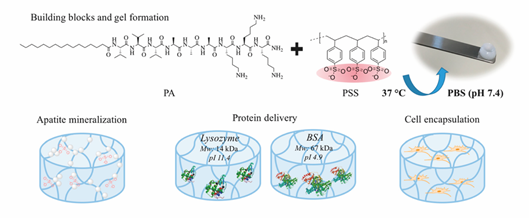
Figure 5. Representative multicomponent supramolecular hydrogel self-assembled between PA and PSS. The PA/PSS hydrogel is multifunctional providing sites for calcium phosphate mineral nucleation and a hydrated network for protein delivery and 3D cell encapsulation. Adapted with permission from [61] Copyright © (2019), American Chemical Society.
The self-assembly of nucleopeptides with single-stranded DNAs (ssDNAs) into hydrogels at physiological pH (pH 7.4) has been reported by Xu and colleagues [62]. To enable interactions between the nucleopeptide and ssDNA, three nucleobases (two thymines and one cytosine) were coupled to ε-amine on the lysine side chains of the peptide Nap-FFKGKGL-OH. The nucleotide formed a weak nanofiber gel on its own, but the addition of ssDNAs induced nanofiber bundling and contributed to the formation of a stronger hydrogel.
An injectable hybrid hydrogel fabricated by an amphiphilic small peptide (Fmoc-FF-OH) and a fullerene derivative called C60 pyrrolidine tris-acid (C60-PTC) was reported. Fmoc-FF-OH itself could self-assemble into a β-sheet nanofibrous transparent hydrogel. Upon integration of C60-PTC, the β-sheet structure changed to α-helix, which mainly resulted from hydrogen bonding together with electrostatic repulsion between Fmoc-FF-OH and C60-PTC. C60-PTC appeared as uniform nanoparticles with diameter of 6 nm instead of the aggregates (110 nm) as observed in water, indicating that the hydrogen bonding and electrostatic repulsion between Fmoc-FF-OH and C60-PTC inhibited the hydrophobic and π-π interactions between C60-PTC molecules. The broadened bands from UV-vis absorption spectra suggested that hydrophobic and π-π interactions between C60-PTC also contributed to the hydrogel formation. As a result, mechanical properties were improved and the 1O2 generation activity of fullerene was enhanced due to the uniform distributed C60-PTC. This led to enhanced wound healing due to the antibacterial effect of sustained reactive oxygen species production.
References
- Langer, R.; Vacanti, J.P. Tissue engineering. Science (New York, NY) 1993, 260, 920–926.
- Caballero Aguilar, L.M.; Silva, S.M.; Moulton, S.E. Growth factor delivery: Defining the next generation platforms for tissue engineering. J. Control. Release Off. J. Control. Release Soc. 2019, 306, 40–58.
- Lee, K.; Silva, E.A.; Mooney, D.J. Growth factor delivery-based tissue engineering: General approaches and a review of re-cent developments. J. R. Soc. Interface 2011, 8, 153–170.
- Rode, M.P.; Batti Angulski, A.B.; Gomes, F.A.; da Silva, M.M.; Jeremias, T.D.S.; de Carvalho, R.G.; Calloni, G.W. Carrageenan hydrogel as a scaffold for skin-derived multipotent stromal cells delivery. J. Biomater. Appl. 2018, 33, 422–434.
- Miranda, D.G.; Malmonge, S.M.; Campos, D.M.; Attik, N.G.; Grosgogeat, B.; Gritsch, K. A chitosan-hyaluronic acid hydrogel scaffold for periodontal tissue engineering. J. Biomed. Mater. Res. Part. B Appl. Biomater. 2016, 104, 1691–1702.
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071.
- Dong, R.; Zhao, X.; Guo, B.; Ma, P.X. Self-Healing Conductive Injectable Hydrogels with Antibacterial Activity as Cell De-livery Carrier for Cardiac Cell Therapy. ACS Appl. Mater. Interfaces 2016, 8, 17138–17150.
- Wang, H.; Heilshorn, S.C. Adaptable hydrogel networks with reversible linkages for tissue engineering. Adv. Mater. (Deer-field Beach, Fla) 2015, 27, 3717–3736.
- Rosales, A.M.; Anseth, K.S. The design of reversible hydrogels to capture extracellular matrix dynamics. Nat. Rev. Mater. 2016, 1, 15012.
- Chen, J.X.; Cao, L.J.; Shi, Y.; Wang, P.; Chen, J.H. In situ supramolecular hydrogel based on hyaluronic acid and dextran de-rivatives as cell scaffold. J. Biomed. Mater. Res. Part A 2016, 104, 2263–2270.
- Dang, I.A.; Kousar, A.; Liu, J.; Mukwaya, V.; Zhao, C.; Wang, F.; Feng, C.L. Mechanically Stable C2-Phenylalanine Hybrid Hydrogels for Manipulating Cell Adhesion. ACS Appl. Mater. Interfaces 2019, 11, 28657–28664.
- Bhattarai, N.; Gunn, J.; Zhang, M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv. Drug Deliv. Rev. 2010, 62, 83–99.
- Wahid, F.; Zhou, Y.N.; Wang, H.S.; Wan, T.; Zhong, C.; Chu, L.Q. Injectable self-healing carboxymethyl chitosan-zinc su-pramolecular hydrogels and their antibacterial activity. Int. J. Biol. Macromol. 2018, 114, 1233–1239.
- Chandramohan, Y.; Jeganathan, K.; Sivanesan, S.; Koka, P.; Amritha, T.M.S.; Vimalraj, S.; Dhanasekaran, A. Assessment of human ovarian follicular fluid derived mesenchymal stem cells in chitosan/PCL/Zn scaffold for bone tissue regeneration. Life Sci. 2021, 264, 118502.
- O’Connor, J.P.; Kanjilal, D.; Teitelbaum, M.; Lin, S.S.; Cottrell, J.A. Zinc as a Therapeutic Agent in Bone Regeneration. Mate-rials (Basel, Switzerland) 2020, 13, 2211.
- Rodell, C.B.; Kaminski, A.L.; Burdick, J.A. Rational design of network properties in guest-host assembled and shear-thinning hyaluronic acid hydrogels. Biomacromolecules 2013, 14, 4125–4134.
- Rosales, A.M.; Rodell, C.B.; Chen, M.H.; Morrow, M.G.; Anseth, K.S.; Burdick, J.A. Reversible Control of Network Properties in Azobenzene-Containing Hyaluronic Acid-Based Hydrogels. Bioconjugate Chem. 2018, 29, 905–913.
- Montanari, E.; Zoratto, N.; Mosca, L.; Cervoni, L.; Lallana, E.; Angelini, R.; Matricardi, P. Halting hyaluronidase activity with hyaluronan-based nanohydrogels: Development of versatile injectable formulations. Carbohydr. Polym. 2019, 221, 209–220.
- Montanari, E.; Di Meo, C.; Coviello, T.; Gueguen, V.; Pavon-Djavid, G. Intracellular Delivery of Natural Antioxidants via Hyaluronan Nanohydrogels. Pharmaceutics 2019, 11, 532.
- Lee, F.; Chung, J.E.; Kurisawa, M. An injectable hyaluronic acid-tyramine hydrogel system for protein delivery. J. Control. Release Off. J. Control. Release Soc. 2009, 134, 186–193.
- Redondo-Gómez, C.; Abdouni, Y.; Becer, C.R.; Mata, A. Self-Assembling Hydrogels Based on a Complementary Host-Guest Peptide Amphiphile Pair. Biomacromolecules 2019, 20, 2276–2285.
- Van de Manakker, F.; van der Pot, M.; Vermonden, T.; van Nostrum, C.F.; Hennink, W.E. Self-Assembling Hydrogels Based on β-Cyclodextrin/Cholesterol Inclusion Complexes. Macromolecules 2008, 41, 1766–1773.
- Van de Manakker, F.; Braeckmans, K.; Morabit, N.E.; De Smedt, S.C.; van Nostrum, C.F.; Hennink, W.E. Protein-Release Behavior of Self-Assembled PEG–b-Cyclodextrin/PEG–Cholesterol Hydrogels. Adv. Funct. Mater. 2009, 19, 2992–3001.
- Li, J. Self-assembled supramolecular hydrogels based on polymer–cyclodextrin inclusion complexes for drug delivery. NPG Asia Mater. 2010, 2, 112–118.
- Harada, A.; Li, J.; Kamachi, M. The molecular necklace: A rotaxane containing many threaded α-cyclodextrins. Nature 1992, 356, 325–327.
- Khodaverdi, E.; Heidari, Z.; Tabassi, S.A.; Tafaghodi, M.; Alibolandi, M.; Tekie, F.S.; Hadizadeh, F. Injectable supramolecular hydrogel from insulin-loaded triblock PCL-PEG-PCL copolymer and γ-cyclodextrin with sustained-release property. AAPS Pharmscitech 2015, 16, 140–149.
- Wu, H.; Song, L.; Chen, L.; Zhang, W.; Chen; Y.; Zang, F.; Zhang, Y. Injectable magnetic supramolecular hydrogel with magnetocaloric liquid-conformal property prevents post-operative recurrence in a breast cancer model. Acta Biomater. 2018, 74, 302–311.
- Xue, B.; Qin, M.; Wu, J.; Luo, D.; Jiang, Q.; Li, Y.; Wang, W. Electroresponsive Supramolecular Graphene Oxide Hydrogels for Active Bacteria Adsorption and Removal. ACS Appl. Mater. Interfaces 2016, 8, 15120–15127.
- Vermonden, T.; Censi, R.; Hennink, W.E. Hydrogels for protein delivery. Chem. Rev. 2012, 112, 2853–2888.
- He, C.; Kim, S.W.; Lee, D.S. In situ gelling stimuli-sensitive block copolymer hydrogels for drug delivery. J. Control. Release Off. J. Control. Release Soc. 2008, 127, 189–207.
- Sanna, D.; Alzari, V.; Nuvoli, D.; Nuvoli, L.; Rassu, M.; Sanna, V.; Mariani, A. β-Cyclodextrin-based supramolecular poly(N-isopropylacrylamide) hydrogels prepared by frontal polymerization. Carbohydr. Polym. 2017, 166, 249–255.
- Song, X.; Zhang, Z.; Zhu, J. Thermoresponsive Hydrogel Induced by Dual Supramolecular Assemblies and Its Controlled Release Property for Enhanced Anticancer Drug Delivery. Biomacromolecules 2020, 21, 1516–1527.
- Zou, H.; Guo, W.; Yuan, W. Supramolecular hydrogels from inclusion complexation of alpha-cyclodextrin with densely grafted chains in micelles for controlled drug and protein release. J. Mater. Chem. B 2013, 1, 6235–6244.
- Bastings, M.M.; Koudstaal, S.; Kieltyka, R.E.; Nakano, Y.; Pape, A.C.; Feyen, D.A.; Dankers, P.Y. A fast pH-switchable and self-healing supramolecular hydrogel carrier for guided, local catheter injection in the infarcted myocardium. Adv. Healthc. Mater. 2014, 3, 70–78.
- Zhang, S.; Bellinger, A.M.; Glettig, D.L.; Barman, R.; Lee, Y.A.; Zhu, J.; Traverso, G. A pH-responsive supramolecular poly-mer gel as an enteric elastomer for use in gastric devices. Nat. Mater. 2015, 14, 1065–1071.
- Tamesue, S.; Takashima, Y.; Yamaguchi, H.; Shinkai, S.; Harada, A. Photoswitchable Supramolecular Hydrogels Formed by Cyclodextrins and Azobenzene Polymers. Angew. Chem. Int. Ed. 2010, 49, 7461–7464.
- Mandl, G.A.; Rojas-Gutierrez, P.A.; Capobianco, J.A. A NIR-responsive azobenzene-based supramolecular hydrogel using upconverting nanoparticles. Chem. Commun. (Cambridge, England) 2018, 54, 5847–5850.
- Mehrban, N.; Abelardo, E.; Wasmuth, A.; Hudson, K.L.; Mullen, L.M.; Thomson, A.R.; Woolfson, D.N. Assessing cellular response to functionalized α-helical peptide hydrogels. Adv. Healthc. Mater. 2014, 3, 1387–1391.
- Zhang, S.; Holmes, T.; Lockshin, C.; Rich, A. Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc. Natl. Acad. Sci. USA 1993, 90, 3334–3338.
- Koutsopoulos, S.; Zhang, S. Two-layered injectable self-assembling peptide scaffold hydrogels for long-term sustained re-lease of human antibodies. J. Control. Release Off. J. Control. Release Soc. 2012, 160, 451–458.
- Clarke, D.E.; Parmenter, C.D.J.; Scherman, O.A. Tunable Pentapeptide Self-Assembled β-Sheet Hydrogels. Angew. Chem. Int. Ed. 2018, 57, 7709–7713.
- Dasgupta, A.; Das, D. Designer Peptide Amphiphiles: Self-Assembly to Applications. Langmuir ACS J. Surf. Colloids 2019, 35, 10704–10724.
- Ghosh, G.; Barman, R.; Sarkar, J.; Ghosh, S. pH-Responsive Biocompatible Supramolecular Peptide Hydrogel. J. Phys. Chem. B 2019, 123, 5909–5915.
- Dehsorkhi, A.; Castelletto, V.; Hamley, I.W. Self-assembling amphiphilic peptides. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2014, 20, 453–467.
- Cui, H.; Webber, M.J.; Stupp, S.I. Self-assembly of peptide amphiphiles: From molecules to nanostructures to biomaterials. Biopolymers 2010, 94, 1–18.
- Zhao, X.; Liu, X.; Zhang, P.; Liu, Y.; Ran, W.; Cai, Y.; Li, Y. Injectable peptide hydrogel as intraperitoneal triptolide depot for the treatment of orthotopic hepatocellular carcinoma. Acta Pharm. Sin. B 2019, 9, 1050–1060.
- Freeman, R.; Han, M.; Álvarez, Z.; Lewis, J.A.; Wester, J.R.; Stephanopoulos, N.; Stupp, S.I. Reversible self-assembly of su-perstructured networks. Science (New York, NY) 2018, 362, 808–813.
- Gačanin, J.; Synatschke, C.V.; Weil, T. Biomedical Applications of DNA-based hydrogels. Adv. Funct. Mater. 2020, 30, 1906253.
- Shao, Y.; Jia, H.; Cao, T.; Liu, D. Supramolecular Hydrogels Based on DNA Self-Assembly. Acc. Chem Res. 2017, 50, 659–668.
- Li, X.; Kuang, Y.; Lin, H.C.; Gao, Y.; Shi, J.; Xu, B. Supramolecular nanofibers and hydrogels of nucleopeptides. Angewandte Chemie (International ed in English) 2011, 50, 9365–9369.
- Baek, K.; Noblett, A.D.; Ren, P.; Suggs, L.J. Design and Characterization of Nucleopeptides for Hydrogel Self Assembly. ACS Appl. Mater. Interfaces 2019, 2, 2812–2821.
- Daly, M.L.; Gao, Y.; Freeman, R. Encoding Reversible Hierarchical Structures with Supramolecular Peptide-DNA Materials. Bioconjugate Chem. 2019, 30, 1864–1869.
- Boback, K.; Bacchi, K.; O’Neill, S.; Brown, S.; Dorsainvil, J. Impact of C-Terminal Chemistry on Self-Assembled Morphology of Guanosine Containing Nucleopeptides. Molecules (Basel, Switzerland) 2020, 25, 5493.
- Li, X.; Kuang, Y.; Shi, J.; Gao, Y.; Lin, H.C.; Xu, B. Multifunctional, biocompatible supramolecular hydrogelators consist only of nucleobase, amino acid, and glycoside. J. Am. Chem. Soc. 2011, 133, 17513–17518.
- Radvar, E.; Azevedo, H.S. Supramolecular Peptide/Polymer Hybrid Hydrogels for Biomedical Applications. Macromol. Bi-osci. 2019, 19, e1800221.
- Xing, R.; Li, S.; Zhang, N.; Shen, G.; Möhwald, H.; Yan, X. Self-Assembled Injectable Peptide Hydrogels Capable of Trigger-ing Antitumor Immune Response. Biomacromolecules 2017, 18, 3514–3523.
- Zhang, Y.; Zhang, H.; Zou, Q.; Xing, R.; Jiao, T.; Yan, X. An injectable dipeptide-fullerene supramolecular hydrogel for pho-todynamic antibacterial therapy. J. Mater. Chem. B 2018, 6, 7335–7342.
- Rajangam, K.; Behanna, H.A.; Hui, M.J.; Han, X.; Hulvat, J.F.; Lomasney, J.W.; Stupp, S.I. Heparin binding nanostructures to promote growth of blood vessels. Nano Lett. 2006, 6, 2086–2090.
- Borges, J.; Sousa, M.P.; Cinar, G.; Caridade, S.G.; Guler, M.O.; Mano, J.F. Nanoengineering Hybrid Supramolecular Multi-layered Biomaterials Using Polysaccharides and Self-Assembling Peptide Amphiphiles. Adv. Funct. Mater. 2017, 27, 1605122.
- Damanik, F.F.R.; Brunelli, M.; Pastorino, L.; Ruggiero, C.; van Blitterswijk, C.; Rotmans, J.; Moroni, L. Sustained delivery of growth factors with high loading efficiency in a layer by layer assembly. Biomater. Sci. 2019, 8,174–188.
- Radvar, E.; Azevedo, H.S. Supramolecular Nanofibrous Peptide/Polymer Hydrogels for the Multiplexing of Bioactive Sig-nals. ACS Biomater. Sci. Eng. 2019, 5, 4646–4656.
- Du, X.; Zhou, J.; Li, X.; Xu, B. Self-assembly of nucleopeptides to interact with DNAs. Interface Focus 2017, 7, 20160116.



