 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Elisabetta Tabolacci | + 4359 word(s) | 4359 | 2021-03-03 09:19:09 | | | |
| 2 | Karina Chen | Meta information modification | 4359 | 2021-03-04 10:07:52 | | | | |
| 3 | Karina Chen | Meta information modification | 4359 | 2021-03-04 10:09:55 | | |
Video Upload Options
Among the inherited causes of intellectual disability and autism, Fragile X syndrome (FXS) is the most frequent form, for which there is currently no cure. In most FXS patients, the FMR1 gene is epigenetically inactivated following the expansion over 200 triplets of a CGG repeat (FM: full mutation). FMR1 encodes the Fragile X Mental Retardation Protein (FMRP), which binds several mRNAs, mainly in the brain. When the FM becomes methylated at 10–12 weeks of gesta-tion, the FMR1 gene is transcriptionally silent.
1. Transcription at the FMR1 Locus and R-Loops
The fine epigenetic control exerted on the FMR1 gene is indicative of a locus exposed to a complex transcriptional regulation. Most of our genome is not translated into proteins, while noncoding RNA (ncRNA) transcripts are particularly abundant in the brain [1]. Some of these are about 200 nucleotides long (lncRNAs: long ncRNAs) and are involved in the epigenetic control of the locus from which they arise. The transcription of lncRNAs may occur from both strands. Sense and antisense transcripts are also described for the FMR1 locus. The most abundant antisense transcript, known as FMR1-AS1, is not expressed in FXS cells and is overexpressed in PM cells, exactly like the sense transcript [2]. FMR1-AS1, consistently expressed at high levels in the brains of unaffected individuals, undergoes alternative splicing, polyadenylation, and exportation to the cytoplasm. Its transcription arises from two alternative promoters: one overlaps with the FMR1 bidirectional promoter and the other is mapped in the second intron of the FMR1 gene. Notably, the latter promoter is mostly used by PM and UFM cells, and it consequently spans the CGG expansion [3]. Furthermore, a region of 9.7 kb corresponding to intron 1 of the FMR1 gene is spliced in normal and PM alleles using a non-consensus CT to AC splicing site. A specific alternative splicing of intron 2, also demonstrated in PM alleles, arises from a non-consensus CT–AC site. Interestingly, a homologous transcript to human FMR1-AS1 is present in the mouse genome, suggesting a conserved cellular function for the antisense transcript [2].
An additional ncRNA, known as FMR4, was identified by Khalil and colleagues. FMR4 arises in the antisense orientation with respect to the FMR1 locus and is particularly expressed in adult frontal cortex and hippocampus. Again, its expression is similar to that of FMR1 mRNA: FXS alleles do not transcribe FMR4, while PM alleles present slight overexpression compared to normal levels of transcription. Transfection experiments were performed in HEK293 cells to functionally correlate both transcripts: siRNAs against the FMR4 transcript and against FMR1 mRNA were transfected to downregulate transcription, while vectors that included their sequence were employed to overexpress them. None of these transfection experiments showed modifications of expression of the other transcript and/or functional correlation between both transcripts [4].
FMR5 and FMR6 represent two additional ncRNAs arising from the FMR1 locus [5]. FMR5 is a lncRNA transcribed 1 kb upstream of the FMR1 transcription start site in the sense orientation. Its transcriptional start site maps in the methylated region upstream of the methylation boundary. It is expressed in several human brain areas of unaffected and FXS individuals, as well as PM carriers. FMR6 is transcribed from the antisense strand overlapping with the 3′ UTR and the last three exons of the FMR1 gene. It is silenced in FXS and PM carriers. Moreover, FMR6 splices the two introns present between the exons 15–16 and 16–17, as for FMR1 mRNA. The splicing mechanism recognizes non-canonical consensus sites, as documented for the splicing of FMR1-AS1[3].
The antisense transcripts may represent intermediaries in the epigenetic regulation of the FMR1 transcription, though their exact role has not yet been clarified. For other loci lncRNAs or antisense transcripts induce the assembly of proteins involved in heterochromatin organization or direct the recruitment of silencing machinery [48]. One could hypothesize that Polycomb group proteins (PcGs) are targeted to the CGG repeat of the FMR1 gene through lncRNAs arising from the locus itself, acting either in cis, as demonstrated for the Kcnq1-Kcnq1ot1 gene cluster [6], or in trans, as described for the Hox-HOTAIR gene clusters [7]. This hypothesis is supported by the evidence that most PcG proteins target G–C-rich regions, so the CGG•CCG-repeats of the FMR1 gene may be silenced by these complexes.
Recently, it has been demonstrated that specific ncRNAs bind to DNMT1; these RNAs have been defined as DNMT1-interacting RNAs, depicting a new class of transcripts [8]. A genome-wide study that led to the identification of these RNAs started from the molecular analysis of the CEBPA gene, involved in hematological malignancies. The DNA methylation levels of the CEBPA locus and the transcriptional levels of an lncRNA of CEBPA, known as ecCEBPA (extra coding CEBPA), are inversely correlated. It was demonstrated that ecCEBPA transcript interacts with DNMT1, preventing CEBPA methylation and promoting CEPBA-mRNA transcription. This functional DNMT1–RNA interaction involves several loci, including FMR1 [8]. These results suggest that DNMT1 is sequestered by RNAs that act as a shield, preventing the methylation of the locus by DNMT1. To propose a targeted therapeutic approach for FXS it will be relevant to focus our attention on understanding interactions between epigenetic enzymes, such as DNMT1, and transcripts stemming from the FMR1 locus. Understanding the mechanisms that regulate the interaction between FMR1 locus and DNMT1 could uncover new targets for potential treatments. A representative model of DNMT1 interaction with DNA and transcription at the FMR1 locus is reported in Figure 1.
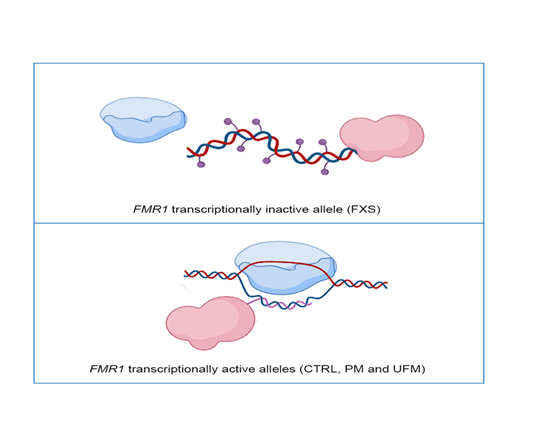
Figure 1. Representative model of DNA methyltransferase 1 (DNMT1) interaction with the FMR1 locus: In FMR1 transcriptionally inactive allele, as in Fragile X syndrome (FXS) individuals, DNMT1 (in pink) freely binds DNA and methylates it (violet dots), preventing transcription through RNA polymerase (in blue) (top panel). In FMR1 transcriptionally active alleles, namely CTRL, PM and UFM, RNA polymerase transcribes RNA (pink line) and DNMT1 binds to nascent RNA and R-loop structures, preventing DNA methylation (bottom panel). Created with BioRender.com.
Recently, Cheung and colleagues showed that RNA:DNA hybrid formation promotes transcription by preventing methylation-induced silencing. The authors studied the BAMBI gene, which is a negative regulator of transforming growth factor β (TGF-β). They observed a decrease in R-loop formation that exposes the BAMBI promoter to the DNA methylation induced by DNMT1. After BAMBI silencing, the TGF-β signaling pathway is activated [9]. When the transcript anneals during or after transcription to the template DNA of its genomic locus of origin to form an RNA:DNA hybrid, the non-template single-stranded DNA (ssDNA) is displaced. These atypical structures with three strands of nucleic acids are called R-loops, and they are most abundant in genomic regions enriched in repetitive elements. Genome-wide methylation data showed that the levels of DNA methylation near human promoter regions and transcriptional levels are directly correlated [10][11]. These findings suggest that the start of transcription at CGI promoters is critical for preventing DNA methylation, although the exact mechanism(s) by which this protection takes place remains unclear [12]. Recent data showed that R-loops can have an epigenetic positive role in the FMR1 locus [13]. R-loops typically originate from guanine-rich (G-rich) regions during gene transcription; they form when an RNA strand invades double-stranded DNA and anneals to the antisense template DNA strand. This causes a Watson–Crick RNA:DNA hybrid and displaces the single stranded DNA (ssDNA) that is not hybridized to RNA [14]. R-loop formation is affected by three main factors—a high density of G, negative supercoiling, and DNA nicks [15]—that influence each other and are not mutually exclusive. DNA:RNA hybridization between nascent RNA and template DNA strand may be favored by both G-clusters and nicks downstream from the promoter on the non-template strand, whereas high G density and negative supercoiling may enhance RNA:DNA hybrid extension and stabilization after its formation [16]. At a reduced G density, R-loop elongation becomes less favorable, leading to the termination of the structure [17]. R-loops are abundant within human gene promoters and terminators, where RNA transcription and processing occur [13][18]. Several studies have demonstrated that an excess or lack of R-loops leads to pathogenic conditions, such as immunodeficiency diseases and neurodegenerative disorders [10][19][20]. Interestingly, it was demonstrated that CGG expansions at the FMR1 locus are associated with an increase of R-loop formation detected by a S9.6 anti-RNA:DNA hybrid antibody that has the ability to specifically bind R-loops. Through a DOX-inducible episomal FMR1 vector, it was secondarily demonstrated that R-loop formation is directly proportional to transcriptional activity. Taken together, these experiments support the assumption that there is a direct correlation between CGG repeat expansions and R-loop formation at the FMR1 locus [21]. Recently, it was also demonstrated that FXS cells accumulate double strand breaks (DSBs), which co-localize with R-loop-forming sequences. Exogenously expressed FMRP in FXS fibroblasts reduces R-loop-induced DSBs, suggesting that FMRP is a genome maintenance protein that prevents R-loop accumulation [22].
2. hESCs and iPSCs as Developmental Model for FXS
To further explore how the lack of FMRP causes the FXS phenotype, several FXS animal models have been developed, such as those of mouse [23], rat [24], zebrafish [25], and fruit fly [26]. Fmr1-knockout models mimic some biochemical, histological, and behavioral findings of FXS patients. Though in vivo studies offer an important experimental tool for characterizing the cellular roles of FMRP, to date there have been no animal models that recapitulate the human process of CGG expansion and methylation. Human embryonic stem cells (hESCs) are a powerful tool in disease modelling thanks to their ability to recapitulate the early stages of human development[27]. However, the availability of FXS hESCs still represents a limiting factor for researchers. Indeed, the origin of hESC is totally dependent on the rare resource of embryos deriving from preimplantation genetic diagnosis (PGD) [28].
hESCs show a unique epigenetic pattern critical for their self-renewal and pluripotency, as they display a largely permissive chromatin configuration with low levels of compact heterochromatin [29]. Furthermore, pluripotency-associated genes, such as Oct4 and Nanog, are mostly unmethylated, and many promoters of developmental transcription factors are characterized by distinctive histone modification signatures that combine active and repressive epigenetic marks. This bivalence guarantees epigenetic plasticity, allowing embryonic stem cells to rapidly regulate gene expression in response to appropriate environmental signals [30]. Similar to pluripotency genes, during differentiation, the fully mutated FMR1 gene undergoes a heterochromatinization process, which involves Polycomb complex, G9a protein, and specific DNA methylation that seems to be guided by a Dicer-mediated siRNA mechanism. Following Dicer cleavage, local methyltransferase SUV39H, which mediates the H3K9me3, is responsible for the heterochromatinization of the FMR1 locus [31]. However, the exact details of the epigenetic mechanism of FMR1 inactivation during development are not yet understood.
Chorionic villi samples from full mutation fetuses were tested for FMR1 expression, and the presence of FMRP was characterized at different stages of gestation [40]. Apparently, FMRP is normally expressed until 13 weeks of gestation, and its expression level is similar to that observed in control fetuses. However, FMRP is unexpressed in fetal tissues although the genetic background of CVS and fetal tissues is identical. These data suggest that the hypermethylation of the FMR1 locus is responsible for the repression of FMR1 transcription and that methylation occurs early in embryogenesis, at around 11 weeks of gestation[32].
In line with these findings, in hESC lines obtained from a preimplantation FXS embryo [31], FMR1 showed normal levels of transcription and translation, which means that the promoter region was unmethylated and exhibited active chromatin features.
Upon the spontaneous differentiation of FXS-hESC lines, a downregulation of FMR1 mRNA level was observed. This decline occurred with the targeted multistep process of heterochromatinization involving firstly histone H3 methylation at lysine 9 or 27 and then DNA methylation [33]. Figure 2 reports the DNA methylation status and the transcriptional activity of the FMR1 locus in hESC and in corresponding differentiated cells. In a recent study [34], FMR1 silencing was monitored during in vitro neuronal differentiation over 60 days. Neurons derived from FXS hESCs showed a progressive decrease of the FMR1 mRNA level until it was totally absent after 51 days post neuronal induction. In particular, in differentiated FXS cells, the FMR1 promoter showed an increase of the repressive H3K9me3 mark, related to loss of the active H3K4me3 mark. Moreover, it was demonstrated that FMR1 mRNA was directly involved in silencing the FMR1 promoter [34]. Briefly, the FMR1 mRNA includes the transcribed CGG-repeat sequence as part of the 5′ UTR and may hybridize with the complementary CGG tract of the FMR1 gene to form an RNA:DNA duplex. Using a small molecule, labeled 1a, the interaction between mRNA and the CGG-repeat portion of the FMR1 gene was found to be disrupted, thus preventing promoter silencing. These results provide a link between trinucleotide-repeat expansion and transcription, as well as a form of RNA-directed gene silencing mediated by RNA:DNA hybrid formation. A direct role of active transcription in the epigenetic silencing of the FMR1 gene was proved by the knockdown of FMR1 mRNA in FXS hESCs, where FMR1 gene is expressed and its promoter region retains histone marks typical of transcriptionally active chromatin. Epigenetic machinery may be triggered at the locus by secondary structures, such as hairpin, formed by CGG repeats in mRNA. The small molecule 1a may unfold and linearize these secondary structures, thus binding the repeating G–G internal loops in the RNA hairpin and inhibiting its thermal melting, preventing FMR1 silencing [34]. However, hESCs employed in this relevant work appear partially methylated, and the mechanism described by the authors does not explain the existence of UFM alleles, which maintain the FMR1 transcription in the presence of CGG expansion in the range of FM.
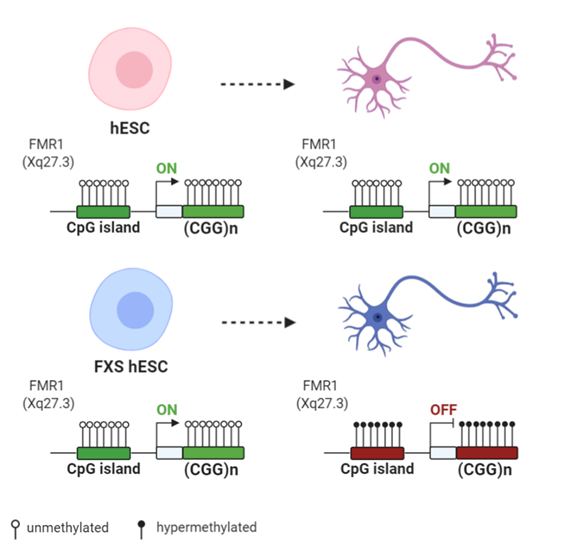
Figure 2. Differences of FMR1 methylation during differentiation of human embryonic stem cells (hESCs) derived from normal and FXS individuals: Both normal and FXS hESC have an unmethylated FMR1 promoter and similar FMR1 expression levels. Upon differentiation, the FMR1 promoter and the CGG expansion become hypermethylated in FXS hESCs, with the subsequent decrease of the FMR1 expression level. Created with BioRender.com.
In the same year, Avitzour et al. (2014) established and characterized various full mutation FXS hESCs lines from affected embryos [35]. In some of the FXS hESCs lines, the FMR1 promoter is partially methylated, ranging from 24% to 65%. These methylation levels remain stable in culture and are directly related to an increase in heterochromatin epigenetic marks. As observed in previous studies [28], in completely unmethylated FXS hESCs, the FMR1 expression level was identical to that of control hESC lines. The results obtained via the bisulfite DNA sequencing of single colonies suggest a wide variability in methylation levels of full mutation FMR1 alleles. It follows that clones among the same cell lines were a mixture of cells that showed either the hypermethylation or hypomethylation of the FMR1 promoter. This is in line with the existence of methylation mosaicism among affected individuals [36]. Furthermore, in FXS hESCs lines, the hypermethylation process, when it occurs, is irreversible, thus leading to a permanent inactivation of FMR1 transcription, whereas in originally unmethylated FXS hESCs lines, FMR1 is repressed during differentiation.
Further support to these findings came from the study of other FXS hESC lines containing a combination of larger methylated and smaller unmethylated full mutation alleles. However, after extended propagation in culture, it was observed that the unmethylated alleles were totally methylated and no FMR1 mRNA was produced [37].
An alternative and valid approach to modelling early stages of FXS pathogenesis is represented by iPSC lines. Though iPSCs closely resemble embryo-derived hESCs, the FMR1 gene remains inactive and shows histone modifications typical of inactive heterochromatin. This observation further confirms that FMR1 silencing occurs early during embryogenesis, and it highlights the epigenetic differences between ESCs and iPSCs in FXS modelling [38][39]. While FXS hESCs can recapitulate early embryonic developmental stages, FXS iPSC lines are a useful model to characterize the neuronal defects found in FXS patients and to identify promising new drugs [39].
If the expansion of >200 CGGs represents the main condition triggering FMR1 gene methylation and silencing, how can the existence of UFM carriers be explained? Considering that FXS patients produce transcripts and R-loops at the first stage of development when the transcription is still active, which are the differences in epigenetic regulation between FXS and UFM individuals? A recent study conducted on iPSCs derived from two unrelated UFM individuals showed that lack of methylation of the FMR1 promoter is maintained during the neuronal differentiation of these cells. However, FMR1 became silenced in a small proportion of iPSC clones, with CGG expansion exceeding 400 repeats, thus indicating that the threshold for DNA methylation in UFM cells is higher than the classical 200 CGG repeat. The methylation of large CGG expansion (over 400 repeats) suggested that UFM individuals preserve the intrinsic ability to methylate their mutated FMR1 gene, but they require larger CGG repeat expansions than those observed in FXS patients. Vice versa, very few iPSC clones derived from FXS patients became unmethylated and transcriptionally active at the FMR1 locus [40]. In another study, reprogramming iPS cells derived from the fibroblasts of a different UFM individual resulted in a methylated and silenced FMR1 gene, possibly due to an artefact of the technical procedure [41].
Recent works employed the CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR associated protein 9) system to reactivate FMR1 transcription, selectively targeting the epigenetic modifications at the FMR1 locus [42][43][44][45]. Two studies edited the CGG repeats in FXS hESCs and iPSCs. In the first study, an sgRNA (single guide RNA), complementary to the DNA target located at the 5′ UTR of the FMR1 gene, generated a single double strand break (DBS) [42]. In the second study, sgRNA oligos were designed to target either side of the CGG repeat tract of the FMR1 gene, resulting in two flanking DSBs [43]. In both cases, the cut site was repaired through non-homologous end joining (NHEJ) activity induced by the Cas9 nuclease. As a result, undifferentiated cells showed a CGG repeat deletion that was followed by the demethylation of the FMR1 promoter, the upregulation of active histone marks, and the expression of the gene. These changes were also maintained throughout differentiation into mature neurons [43]. Elsewhere, instead of DSB-based editing, a methylation editing approach was used to reverse the excessive methylation of CGG repeats in the FMR1 gene [44]. In this work, an FXS iPSC model was used to recapitulate the hypermethylation of CGG repeat expansion and epigenetic silencing of FMR1. The results showed that the specifically targeted demethylation of CGG repeats by fusing the Cas9 nuclease to the catalytic domain of the TET DNA demethylase was sufficient to reactivate FMR1 and to partially recover FMRP expression. Upon in vitro differentiation, the reactivation of FMR1 was maintained in edited neurons, which displayed a normal FMR1 expression and restored electrophysiological properties [44]. Haenfler et al. (2018) used an alternative strategy to reactivate FMR1 based on transient transfection. The authors combined the Cas9 nuclease with VP16 transcriptional activation domain in order to robustly enhance FMR1 transcription and increase FMRP levels [45] (Figure 3).
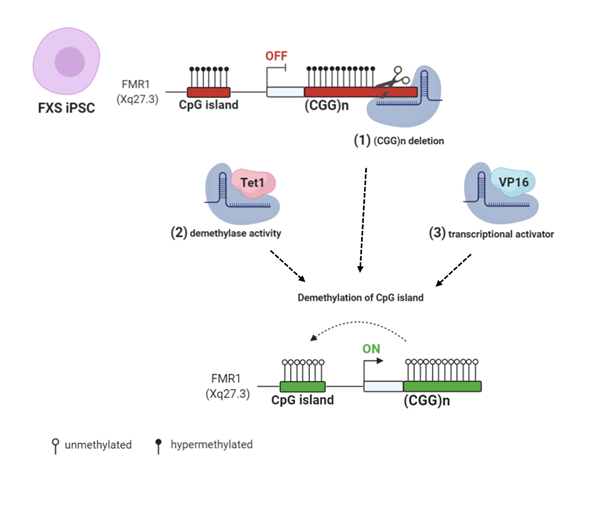
Figure 3. CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR associated protein 9) genome and epigenome editing strategies: (1) FXS iPSCs were edited with CRISPR/Cas9 to remove the CGG repeats in the FMR1 gene [84,85]. (2) Targeted demethylation of CGG repeats was achieved by Cas9-Tet1 [86]. (3) Transcriptional activator VP16 fused to Cas9 robustly enhanced FMR1 transcription [45]. All these CRISPR/Cas9 strategies resulted in FMR1 transcription and Fragile X Mental Retardation Protein (FMRP) production by edited FXS cells. Created with BioRender.com.
Taken together, these results suggest that dynamic epigenetic changes in undifferentiated cells demonstrate the possibility of using multiple complementary strategies to obtain the reactivation of the FMR1 locus. Moreover, the CRISPR/Cas9 editing system could eventually lead to a therapeutic approach in patients with FXS, albeit with caveats regarding the safety and accuracy of this method.
3. Epigenetic Approach to Treat FXS
Two different approaches can be proposed to treat FXS: (1) to compensate for the lack of FMRP, one can intervene on pathways in which the protein is involved; (2) to reactivate the expression of the otherwise intact FMR1 gene, one can act on the epigenetic mechanisms causing its transcriptional inactivation. In CNS cells, and specifically at the synapses, FMRP mainly acts as protein synthesis repressor. The effect of a lack of FMRP on mGLuR signaling [46] has prompted several clinical trials with the purpose of compensating for the lack of FMRP. A promising trial was carried out with AFQ056, a selective inhibitor of mGluR5 with a presumptive epigenetic effect [47][48]. However, a subsequent larger trial was discontinued due to a lack of confirmatory evidence. The existence of the rare UFM individuals, in which CGG expansion remains unmethylated and transcriptionally active, suggested an alternative epigenetic strategy to cure FXS by restoring FMR1 expression. The epigenetic approach is based on the plasticity of epigenetic signatures, which may be reverted from heterochromatin markers in FXS-silenced alleles to more permissive markers typically described in transcriptionally active cells. Cytosines can be demethylated using 5-azacytidine (5-azaC) or, more efficiently and specifically, 5-aza-2-deoxycytidine (5-azadC). This latter compound is an analog of deoxycytidine and is incorporated into DNA during cell divisions blocking DNA methyltransferases, particularly DNMT1 [49]. Our group firstly obtained the in vitro reactivation of the FMR1 gene by treating FXS lymphoblastoid cells with 5-azadC [50]. The reactivation was documented by the reappearance of both FMR1-mRNA and, in a fraction of treated cells, FMRP. The observed discrepancy between mRNA and protein levels could have been due to the lower translational efficiency in presence of CGG expansion. A synergistic effect on FMR1 reactivation was obtained by the combined treatment with 5-azadC and several HDAC inhibitors such as butyrate, phenylbutyrate, and trichostatin A. Treatments with HDAC inhibitors alone did not induce any significant reactivation of the FMR1 gene [51]. Evidence emerging from epigenetic treatments suggests that DNA methylation is prevalent over histone acetylation levels at the FMR1 locus [52], as was also reported for other heavily methylated genes [53]. Additionally, romidepsin and vorinostat, two FDA-approved HDAC inhibitors, have been tested for their ability to reactivate the FMR1 gene. Like other HDAC inhibitors, these drugs weakly reactivate the FMR1 gene in FXS cells; in a few cells, they induce a modest reactivation, while other cell lines exhibit no reactivation. Moreover, most inhibitors of HDAC enzymes are cytotoxic, thus limiting their use in clinical trials for FXS [54].
Treatments of FXS cells with 5-azadC have shown that this compound is able to induce increased histone acetylation and H3K4 methylation levels, while H3K9 methylation is only partially reduced [55]. These epigenetic marks are those typical of a euchromatic configuration of the FMR1 promoter, effectively converting a methylated FXS into an UFM. Notably, the effect of 5-azadC on genomic DNA methylation is not indiscriminate; rather, it is restricted almost exclusively to the FMR1 promoter. To support this point, using a microarray screening of 10,814 transcripts, Suzuki et al. (2002) showed that only 51 genes are transcriptionally upregulated after 5-azadC treatment and 23 genes are overexpressed after trichostatin A treatment [56]. We also demonstrated that the reactivating effect of 5-azadC on the mutant FMR1 gene lasts for several weeks in vitro [57]. A relevant criticism resulting from clinical application of 5-azadC is its toxicity. The use of both 5-azaC and 5-azadC in the treatment of hematological malignancies is generally well-tolerated [58], although their effects over a long time period are not known. Another obstacle to the clinical treatment of FXS with 5-azadC is represented by the evidence that this compound requires cell division, while neurons, the main target cells of the treatment, normally do not divide. Interestingly, there is evidence that 5-azadC effectively induces a reduction of the maintenance DNA methyltransferase DNMT1 through proteasomal degradation, requiring minimal incorporation into DNA [59]. Furthermore, the treatment of FXS-iPS cells and their derived neurons with 5-azacytidine causes a robust FMR1 reactivation [60]. Though preliminary, this study demonstrated that a targeted epigenetic intervention is feasible. However, the major concern about the clinical use of these epigenetic drugs, like 5-azadC or HDAC inhibitors, is again derived from their mechanism of action that may be extended to the whole genome with unspecific and off-target effects.
Further experiments have corroborated the feasibility of an epigenetic intervention to reactivate the mutant FMR1 gene. Valproic acid (VPA), an FDA-approved drug to treat seizures and mood disorders, demonstrated a (modest) reactivating effect on FMR1 gene by acting as histone acetylator without a DNA demethylation effect [61]. Clinical trials based on the use of VPA showed a decrease in hyperactivity disorder in FXS patients, thus further encouraging epigenetic intervention in this genetic condition [62]. A significant reduction of hyperactivity disorder was obtained in a previous clinical trial with L-acetylcarnitine (ALC) [63], a natural compound that may directly increase histone acetylation levels by acting as a donor of acetyl groups. Unfortunately, when ALC is used alone, it is not able to reactivate the FMR1 gene in vitro, as demonstrated for other similar compounds [51].
In a recent paper, an in vivo platform of FXS iPSCs and differentiated FXS transplants was developed to screen small-molecules and to quickly evaluate the ability of tested compounds to target FMR1 inactivation [64]. FMR1-reactivating compounds are evaluated for their additive and long-term effects. Higher FMR1 reactivation levels and lower DNA methylation levels of the FMR1 promoter in FXS transplants were obtained by the combined use of 5-azadC and DZNep (an SAH hydrolase inhibitor) than by treatment with 5-azadC alone. Interestingly, a small reduction of FMR1 methylation levels was obtained by the use of DZNep alone, which may indirectly inhibit histone modifications. Unlike the chemicals used in other studies [58][65], the reactivating effect of DZNep appears to be maintained for a prolonged period of time. These combined treatments with different epigenetic compounds could reduce the 5-azadC concentration required for FMR1 reactivation, thus diminishing the potentially toxic side effects of this demethylating agent used in monotherapy. A platform for the analysis of FMR1-reactivating treatments in vivo was also developed on two humanized animal models carrying human FXS-affected cells: one implied the generation of a differentiated human FXS transplant in immunocompromised mice and the second involved the transplantation of a specified population of human FXS neural progenitor cells into mouse brains. In both cell types, 5-azadC administration was able to restore FMR1 expression, representing a proof of principle for FMR1 demethylation in an in vivo model [64]. Lastly, small molecule inhibitors of H3K9 methylation, such as chaetocin, BIX01294, and DZNep significantly delay the re-silencing of 5-azadC-reactivated FMR1 alleles. These results also demonstrated that H3K9 methylation precedes DNA methylation and that the removal of DNA methylation with 5-azadC is necessary to obtain the optimal effect of HMT inhibitors on FMR1 gene expression [66].
References
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al.. Landscape of transcription in human cells. Nature 2012, 489, 101–108.
- Ladd, P.D.; Smith, L.E.; Rabaia, N.A.; Moore, J.M.; Georges, S.A.; Hansen, R.S.; Hagerman, R.J.; Tassone, F.; Tapscott, S.J.; Filippova, G.N. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 2007, 16, 3174–3187.
- Lanni, S.; Goracci, M.; Borrelli, L.; Mancano, G.; Chiurazzi, P.; Moscato, U.; Ferrè, F.; Helmer-Citterich, M.; Tabolacci, E.; Neri, G.. Role of CTCF protein in regulating FMR1 locus transcription. PLoS Genet. 2013, 9, e1003601.
- Khalil, A.; M.; Faghihi, M.A.; Modarresi, F.; Brothers, S.P.; Wahlestedt, C. A novel RNA transcript with antiapoptotic func-tion is silenced in fragile X syndrome. PLoS ONE 2008, 3, e1486.
- Pastori, C.; Peschansky, V.J.; Barbouth, D.; Mehta, A.; Silva, J.P.; Wahlestedt, C. Comprehensive analysis of the transcrip-tional landscape of the human FMR1 gene reveals two new long noncoding RNAs differentially expressed in Fragile X syn-drome and Fragile X-associated tremor/ataxia syndrome. Hum. Genet. 2014, 133, 59–67.
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-Dinardo, D.; Kanduri, C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell. 2008, 32,232-346.
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323.
- Di Ruscio, A.; Ebralidze, A.K.; Benoukraf, T.; Amabile, G.; Goff, L.A.; Terragni, J.; Figueroa, M.E.; Lobo De Figueiredo Pontes, L.; Alberich-Jorda, M.; Zhang, P.; et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 2013,503,371-376.
- Grunseich, C.; Wang, I.X.; Watts, J.A.; Burdick, J.T.; Guber, R.D.; Zhu, Z.; Bruzel, A.; Lanman, T.; Chen, K.; Schindler, A.B.; et al. Senataxin Mutation Reveals How R-Loops Promote Transcription by Blocking DNA Methylation at Gene Promoters. Mol. Cell. 2018, 69426–69437.
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Sung, K.W.K.; Rigoutsos, I.; Loring, J.; et al. Dy-namic changes in the human methylome during differentiation. Genome Res. 2010, 20320–20331.
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462315–22.
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chédin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell. 2012, 45, 814–825.
- Costantino, L.; Koshland, D. The Yin and Yang of R-loop biology. Curr. Opin. Cell Biol. 2015, 34, 39–45.
- Thomas, M.; White, R.L.; Davis, R.W. Hybridization of RNA to double-stranded DNA: Formation of R-loops. Proc. Natl. Acad. Sci. 1976, 73, 2294–2298.
- Roy, D.; Zhang, Z.; Lu, Z.; Hsieh, C.L.; Lieber, M.L. Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: A nick can serve as a strong R-loop initiation site. Mol. Cell Biol. 2010, 30, 146–159.
- Aguilera, A.; Garcia-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124.
- Allison, D.F.; Wang, G.G. R-loops: Formation, function, and relevance to cell stress. Cell Stress. 2019, 21, 38–46.
- Chen, L.; Chen, J.Y.; Zhang, X.; Gu, Y.; Xiao, R.; Shao, C.; Tang, P.; Qian, H.; Luo, D.; Li, H.; et al. R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-loops with Transcriptional Pausing at Gene Promoters. Mol. Cell 2017, 68745–68757.
- Sarkar, K.; Han, S.S.; Wen, K.K.; Ochs, H.D.; Dupré, L.; Seidman, M.M.; Vyas, Y.M. R-loops cause genomic instability in T helper lymphocytes from patients with Wiskott-Aldrich syndrome. J. Allergy Clin. Immunol. 2018, 142219–142234.
- Groh, M.; Lufino, M.M.P.; Wade-Martins, R.; Gromak, N. R-loops associated with triplet repeat expansions promote gene si-lencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 2014, 10, e1004318.
- Loomis, E.W.; Sanz, L.A.; Chédin, F.; Hagerman, P.J. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 2014, 10, e1004294.
- Chakraborty, A.; Jenjaroenpun, P.; Li, J.; El Hilali, S.; McCulley, A.; Haarer, B.; Hoffman, E.A.; Belak, A.; Thorland, A.; Hehnly, H.; et al. Replication Stress Induces Global Chromosome Breakage in the Fragile X Genome. Cell Reports. 2020, 32108179.
- Kazdoba, T.M.; Leach, P.T.; Silverman, J.L.; Crawley, J.N. Modeling fragile X syndrome in the Fmr1 knockout mouse. Intrac-table Rare Dis Res. 2014, 3, 118–133.
- Hamilton, S.M.; Green, J.R.; Veeraragavan, S.; Yuva, L.; McCoy, A.; Wu, Y.; Warren, J.; Little, L.; Ji, D.; Cui, X.; et al. Fmr1 and Nlgn3 knockout rats: Novel tools for investigating autism spectrum disorders. Behav. Neurosci. 2014, 128, 103–109.
- Hu, J.; Chen, L.; Yin, J.; Yin, H.; Huang, Y.; Tian, J. Hyperactivity, Memory Defects, and Craniofacial Abnormalities in Zebrafish fmr1 Mutant Larvae. Behav. Genet. 2020, 50 152-160.
- Drozd, M.; Bardoni, B.; Capovilla, M. Modeling Fragile X Syndrome in. Front. Mol. Neurosci. 2018, 11, 124.
- Prajumwongs, P.; Weeranantanapan, O.; Jaroonwitchawan, T.; Noisa, P. Human Embryonic Stem Cells: A Model for the Study of Neural Development and Neurological Diseases. Stem Cells Int. 2016, 2016, 2958210.
- Eiges, R.; Urbach, A.; Malcov, M.; Frumkin, T.; Schwartz, T.; Amit, A.; Yaron, Y.; Eden, A.; Yanuka, O.; Benvenisty, N.; et al. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically di-agnosed embryos. Cell Stem Cell 2007, 1, 568–577.
- Boland, M.J.; Nazor, K.L.; Loring, J.F. Epigenetic regulation of pluripotency and differentiation. Circ. Res. 2014, 115 311-24.
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125 315-26.9.
- Hecht, M.; Tabib, A.; Kahan, T.; Orlanski, S.; Gropp, M.; Tabach, Y.; Yanuka, O.; Benvenisty, N.; Keshet, I.; Cedar, H. Epi-genetic mechanism of FMR1 inactivation in Fragile X syndrome. Int. J. Dev. Biol. 2017, 61, 285–292.
- Willemsen, R.; Bontekoe, C.J.; Severijnen, L.A.; Oostra, B.A. Timing of the absence of FMR1 expression in full mutation cho-rionic villi. Hum. Genet. 2002, 110, 601–605.
- Grewal, S.I.; Jia, S.; Heterochromatin revisited. Nat. Rev. Genet. 2007, 8, 35–46.
- Colak, D.; Zaninovic, N.; Cohen, M.S.; Rosenwaks, Z.; Yang, W.Y.; Gerhardt, J.; Disney, M.D.; Jaffrey, S.R. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 2014, 343, 1002–1005.
- Avitzour, M.; Mor-Shaked, H.; Yanovsky-Dagan, S.; Aharoni, S.; Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Schonberger, O.; Levy-Lahad, E.; Epsztejn-Litman, S.; et al. FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Reports 2014, 3, 699–706.
- Baker, E.K.; Arpone, M.; Vera, S.A.; Bretherton, L.; Ure, A.; Kraan, C.M.; Bui, M.; Ling, L.; Francis, D.; Hunter, M.F.; et al. Intellectual functioning and behavioural features associated with mosaicism in fragile X syndrome. J. Neurodev. Disord. 2019, 11 41.
- Zhou, Y.; Kumari, D.; Sciascia, N.; Usdin, K. CGG-repeat dynamics and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons. Mol. Autism 2016, 7, 42.
- Urbach, A.; Bar-Nur, O.; Daley, G.Q.; Benvenisty, N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 2010, 6, 407–411.
- Abu Diab, M.; Eiges, R. The Contribution of Pluripotent Stem Cell (PSC)-Based Models to the Study of Fragile X Syndrome (FXS). Brain Sci. 2019, 9, 42.
- Brykczynska, U.; Pecho-Vrieseling, E.; Thiemeyer, A.; Klein, J.; Fruh, I.; Doll, T.; Manneville, C.; Fuchs, S.; Iazeolla, M.; Beibel, M.; et al. CGG Repeat-Induced FMR1 Silencing Depends on the Expansion Size in Human iPSCs and Neurons Carrying Un-methylated Full Mutations. Stem Cell Rep. 2016, 7, 1059–1071.
- de Esch, C.E.; Ghazvini, M.; Loos, F.; Schelling-Kazaryan, N.; Widagdo, W.; Munshi, S.T.; van der Wal, E.; Douben, H.; Gunhanlar, N.; Kushner, S.A.; et al. Epigenetic characterization of the FMR1 promoter in induced pluripotent stem cells from human fibroblasts carrying an unmethylated full mutation. Stem Cell Reports 2014, 3, 548–555.
- Park, C.Y.; Sung, J.J.; Lee, J.S.; Yanuka, O.; Benvenisty, N.; Kim, D.W.. Reversion of FMR1 Methylation and Silencing by Ed-iting the Triplet Repeats in Fragile X iPSC-Derived Neurons. Cell Rep. 2015, 13234–13241.
- Xie, N.; Gong, H.; Suhl, J.A.; Chopra, P.; Wang, T.; Warren, S.T. Reactivation of FMR1 by CRISPR/Cas9-Mediated Deletion of the Expanded CGG-Repeat of the Fragile X Chromosome. PLoS ONE 2016, 11e0165499.
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172979–992.
- Haenfler, J.M.; Skariah, G.; Rodriguez, C.M.; Monteiro da Rocha, A.; Parent, J.M.; Smith, G.D.; Todd, P.K. Targeted Reacti-vation of FMR1 Transcription in Fragile X Syndrome Embryonic Stem Cells. Front. Mol. Neurosci. 2018, 11, 282.
- Bear, M.F.; Huber, K.M.; Warren, S.T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27, 370–377.
- Jacquemont, S.; Curie, A.; des Portes, V.; Torrioli, M.G.; Berry-Kravis, E.; Hagerman, R.J.; Ramos, F.J.; Cornish, K.; He, Y.; Paulding, C.; et al. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci. Transl. Med. 2011, 3, 64ra1.
- Tabolacci, E.; Pirozzi, F.; Gomez-Mancilla, B.; Gasparini, F.; Neri, G. The mGluR5 antagonist AFQ056 does not affect methyl-ation and transcription of the mutant FMR1 gene in vitro. BMC Med. Genet. 2012, 13,13.
- Jackson-Grusby, L.; Laird, P.W.; Magge, S.N.; Moeller, B.J.; Jaenisch, R. Mutagenicity of 5-aza-2'-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc. Natl. Acad. Sci. USA 1997, 94, 4681–4685.
- Chiurazzi, P.; Pomponi, M.G.; Willemsen, R.; Oostra, B.A.; Neri, G. In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum. Mol. Genet. 1998, 7,109-113.
- Chiurazzi, P.; Pomponi, M.G.; Pietrobono, R.; Bakker, C.E.; Neri, G.; Oostra, B.A. Synergistic effect of histone hyperacetyla-tion and DNA demethylation in the reactivation of the FMR1 gene. Hum. Mol. Genet. 1999, 8, 2317–2323.
- Pietrobono, R.; Pomponi, M.G.; Tabolacci, E.; Oostra, B.A.; Chiurazzi, P.; Neri, G. Quantitative analysis of DNA demethyla-tion and transcriptional reactivation of the FMR1 gene in fragile X cells treated with 5-azadeoxycytidine. Nucleic Acids Res. 2002, 30, 3278–3285.
- Cameron, E.E.; Bachman, K.E.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107.
- Dolskiy, A.A.; Pustylnyak, V.O.; Yarushkin, A.A.; Lemskaya, N.A.; Yudkin, D.V. Inhibitors of Histone Deacetylases Are Weak Activators of the FMR1 Gene in Fragile X Syndrome Cell Lines. Biomed. Res. Int. 2017, 35, 826–831.
- Tabolacci, E.; Pietrobono, R.; Moscato, U.; Oostra, B.A.; Chiurazzi, P.; Neri, G. Differential epigenetic modifications in the FMR1 gene of the fragile X syndrome after reactivating pharmacological treatments. Eur. J. Hum. Genet. 2005, 13, 641–648.
- Suzuki, H.; Gabrielson, E.; Chen, W.; Anbazhagan, R.; van Engeland, M.; Weijenberg, M.P.; Herman, J.G.; Baylin, S.B. A ge-nomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat. Genet. 2002, 31, 141–149.
- Tabolacci, E.; Mancano, G.; Lanni, S.; Palumbo, F.; Goracci, M.; Ferrè, F.; Helmer-Citterich, M.; Neri, G. Genome-wide meth-ylation analysis demonstrates that 5-aza-2-deoxycytidine treatment does not cause random DNA demethylation in fragile X syndrome cells. Epigenet. Chromatin 2016, 9, 12.
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013, 33, 2989–2896.
- Ghoshal, K.; Datta, J.; Majumder, S.; Bai, S.; Kutay, H.; Motiwala, T.; Jacob, S.T. 5-Aza-deoxycytidine induces selective deg-radation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology do-main, and nuclear localization signal. Mol. Cell Biol. 2005, 25, 4727–4741.
- Bar-Nur, O.; Caspi, I.; Benvenisty, N. Molecular analysis of FMR1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J. Mol. Cell Biol. 2012, 4, 180–183.
- Tabolacci, E.; De Pascalis, I.; Accadia, M.; Terracciano, A.; Moscato, U.; Chiurazzi, P.; Neri, G. Modest reactivation of the mutant FMR1 gene by valproic acid is accompanied by histone modifications but not DNA demethylation. Pharmacogenet. Genomics 2008, 18, 738–741.
- Torrioli, M.; Vernacotola, S.; Setini, C.; Bevilacqua, F.; Martinelli, D.; Snape, M.;Hutchison, J.A.; Di Raimo, F.R.; Tabolacci, E.; Neri, G. Treatment with valproic acid ameliorates ADHD symptoms in fragile X syndrome boys. Am. J. Med. Genet. A 2010, 152A, 1420–1427.
- Torrioli, M.G.; Vernacotola, S.; Peruzzi, L.; Tabolacci, E.; Mila, M.; Militerni, R.; Musumeci, S.; Ramos, F.J.; Frontera, M.; Sorge, G.; ,et al. A double-blind, parallel, multicenter comparison of L-acetylcarnitine with placebo on the attention deficit hyperactivity disorder in fragile X syndrome boys. Am. J. Med. Genet. A 2008, 146A, 803-812.
- Vershkov, D.; Fainstein, N.; Suissa, S.; Golan-Lev, T.; Ben-Hur, T.; Benvenisty, N. FMR1 Reactivating Treatments in Fragile X iPSC-Derived Neural Progenitors In Vitro and In Vivo. Cell Rep. 2019, 262531–2539.
- Kumari, D.; Usdin, K. Polycomb group complexes are recruited to reactivated FMR1 alleles in Fragile X syndrome in response to FMR1 transcription. Hum. Mol. Genet. 2014, 236575–236583.
- Kumari, D.; Sciascia, N.; Usdin, K. Small Molecules Targeting H3K9 Methylation Prevent Silencing of Reactivated FMR1 Al-leles in Fragile X Syndrome Patient Derived Cells. Genes 2020, 11, 356.

