 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Amelia PINA | + 3283 word(s) | 3283 | 2021-03-03 10:08:56 | | | |
| 2 | Rita Xu | -1653 word(s) | 1630 | 2021-03-03 10:33:51 | | |
Video Upload Options
Plant viruses are obligate parasites that need to usurp plant cell metabolism in order to infect their hosts. Imaging techniques have been used for quite a long time to study plant virus–host interactions, making it possible to have major advances in the knowledge of plant virus infection cycles.
1. Introduction
The obligate parasitic nature of viruses, associated to the small repertoire of proteins encoded by their tiny genomes, makes tight and finely-tuned virus–host interactions indispensable for them. Understanding the mechanisms underlying these interactions may provide better knowledge of the viruses’ biology as well as the fundamental processes of plant physiology, and this may even point to novel means of controlling these harmful pathogens. The successful infection of a plant by a virus involves a few basic steps, which include entry, desencapsidation, translation, replication, cell-to-cell movement, encapsidation, vascular transport, and plant-to-plant transmission, which can be horizontal through vectors or mechanical wounds, and/or vertical through seeds and pollen. No less important is the fact that along these steps, viruses need to counter host defenses, including RNA silencing as well as protein-mediated basal and specific immunities. In the lines below, we briefly review the imaging techniques used to study plant–virus interactions during these steps.
2. Imaging Techniques Used in Plant Virology
2.1. Macroscopic Techniques
Before embarking in a microscopy study, it is fundamental to understand how the virus is distributed in the tissues under observation to precisely define the specimen to be placed under the microscope; for this, the use of a macroscopic technique such as hybridization on tissue-prints may provide optimal results. Tissue-printing hybridization is a simple technique for transferring macromolecules, such as viral nucleic acids, onto positively charged nylon or nitrocellulose membranes blotted directly from plant organ surfaces. The resulting blot is an image of the tissue surface. Initially, immobilized viral target nucleic acids were detected using the hybridization of radiolabeled nucleic acid probes and exposure to X-ray film, but later, digoxigenin-labeled nucleic acid probes combined with chemiluminescent detection became the norm [1] (Figure 1). Tissue-printing hybridization can be used as an initial approach to study the long distance movement of viruses in fully susceptible or partially resistant plants [2][3][4][5][6][7]. Moreover, this technique can be used to colocalize both host mRNAs and viroid or virus RNAs in plant tissues [8][9][10][11][12].
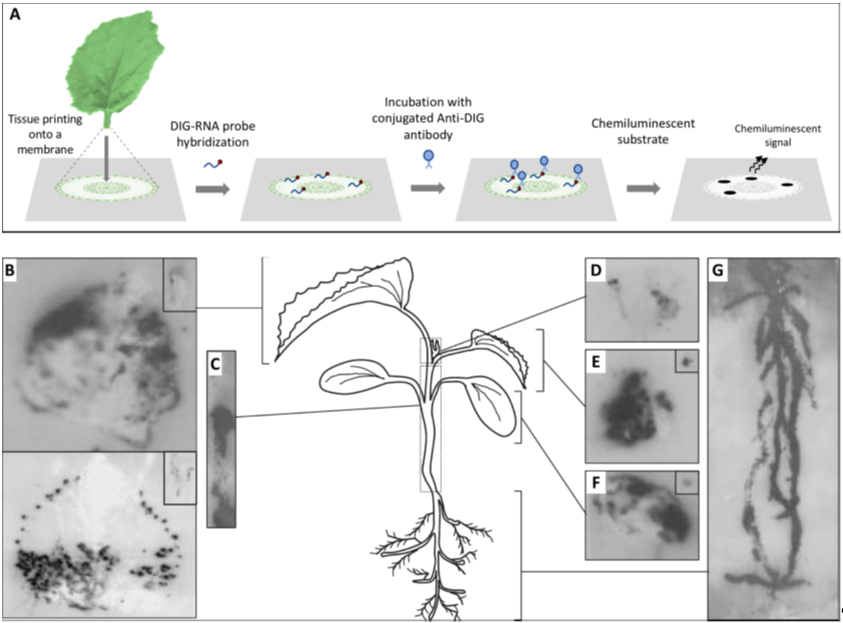
Figure 1. Analysis of the spatial distribution of a virus in infected plants by tissue printing hybridization. (A) Schematic representation of the tissue printing hybridization technique. Frequently after sectioning, the organ surface is printed onto a positively charged nylon or nitrocellulose membrane. Macromolecules, including viral nucleic acids, are transferred from the tissue to the membrane. The blotted membrane is then incubated with a digoxigenin (DIG)-labeled nucleic acid probe and the target nucleic acid is detected after incubation with a conjugated anti-DIG antibody and the appropriated chemiluminescent substrate. (B–G) Tissue print hybridization using a digoxigenin-labelled RNA probe for detecting melon necrotic spot virus (MNSV) vRNA in (B) the first systemic leaves (developed leaves), (C) hypocotyl and main stem, (D) shoot tip, (E) second systemic leaf (young leaf), (F) inoculated cotyledon, and (G) roots of MNSV-infected melon plants. Insets displayed in B, E, and F corresponds to longitudinal or cross-sectional printings of the petioles. This figure is adapted with permission from [8].
2.2. Light Microscopy
Plant tissue is quite difficult to prepare in order to obtain good specimen preservation. The cell wall and large vacuoles present in plant cells cause problems of osmolarity, producing trouble with fixation, dehydration, and embedding. It is important for both light and transmission electron microscopy studies to try different fixation methods, especially for protein or nucleic acid localization experiments. For example, we successfully used combinations of glutaraldehyde plus p-formaldehyde [4][13][14], although p-formaldehyde or formaldehyde alone could also give good results when working with plant tissues [3][15]. Choosing an adequate embedding medium is also important. For plant tissues, paraffin embedding is quite adequate for light microscopy [16], although it is time consuming. Alternatives to chemical fixation could be considered when the required equipment is available, including cryofixation and high pressure freezing [17].
Light microscopy is frequently complemented by the immunodetection of specific host or viral proteins. As with light microscopy, immunodetection can also complement electron microscopy, and both share similar methodological constraints. For light microscopy, specific antibodies are usually detected using a digoxigenin (DIG)-conjugate anti-immunoglobulin secondary antibody, whereas for electron microscopy, antibodies conjugated with gold particles are used (see in next section 2.3). Likewise, in situ hybridization (ISH) provides the means to detect specific plant and viral nucleic acids in plant tissue sections. Similar to immunodetection, ISH can provide cellular resolution and in some cases, subcellular resolution. ISH has been widely used to provide spatial and temporal information on the localization of host and virus nucleic acids [2][4][5][6][13][14][18][19][20]. ISH involves the hybridization of tagged nucleic acid probes to target viral nucleic acids within thin tissue sections followed by the detection of the tagged probe with an anti-tag antibody [21]. The most commonly used probes are short (150–300 nucleotides), in vitro synthesized single-stranded complementary RNAs (cRNAs), or DNAs (cDNAs) labeled with digoxigenin-11-UTP or biotin. Hybridized tagged probes are detected using anti-tag antibodies conjugated to alkaline phosphatase (AP) or horseradish peroxidase (HRP). After incubation with AP- or HRP-appropriate substrates, a chromogenic signal reveals the precise histological distribution of the viral nucleic acid under the light microscope. By combining differentially-targeted probes with different tags, it is possible to colocalize the nucleic acids of different viruses in a tissue section. Alternatively, the observation of consecutive sections, each hybridized with a different probe, may allow for the colocalization of the nucleic acids of different viruses. We used this methodology to colocalize two virus isolates belonging to different strains in mixed infections (Figure 2) [16]. A number of variations to the traditional ISH protocols have been reported, including fluorescence in situ hybridization (FISH) [22][23], whole mount ISH (WISH) [14] and in situ PCR and RT-PCR [24][25][26][27][28][29][30][31], although PCR and RT-PCR have not been widely used in plant virology. WISH is an alternative for the localization of proteins or nucleic acids in small nonsectioned specimens [14] (see below in section 4), but it is not always feasible.
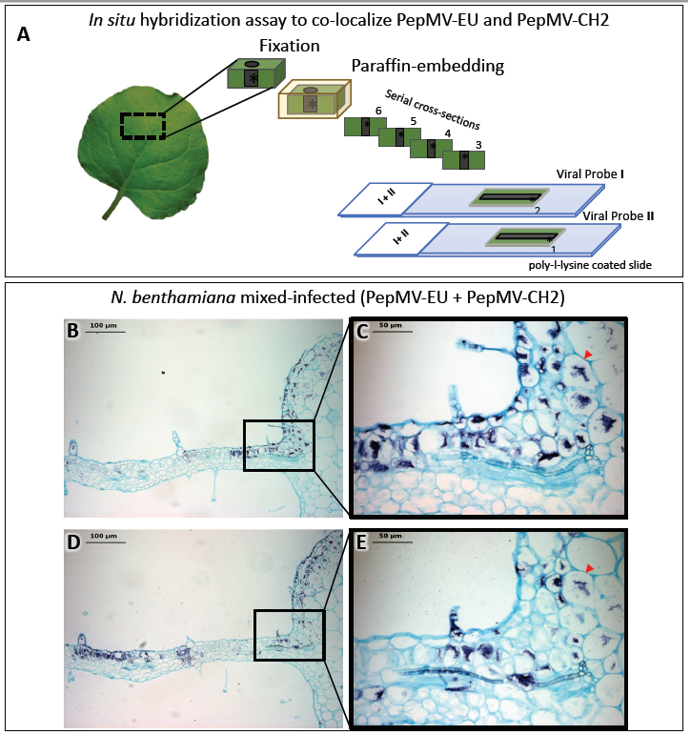
Figure 2. In situ hybridization (ISH) assay on consecutive serial cross sections of Nicotiana benthamiana leaves infected with two isolates belonging to different strains of pepino mosaic virus (PepMV-EU and PepMV-CH2). (A) Schematic representation of the ISH assay. Small pieces of mock and mixed infected N. benthamiana leaves were fixed with paraformaldehyde (PFA), embedded into paraffin and sectioned. The ISH was performed on consecutive leaf cross sections of the same sample by using each probe (I or II) in each slide (1 or 2), respectively. (B, D) Images of the ISH performed on a pair of consecutive leaf serial cross sections from mixed (PepMV-EU + PepMV-CH2) inoculated plants with either the riboprobe for PepMV-EU (B) or PepMV-CH2; both viruses are located in dark-blue colored areas of the leaf sections distributed in patches of infected tissue, mixed with areas of noninfected tissue. (C, E) Higher magnification of the area boxed in B, D, respectively. The viral RNAs of both PepMV-EU and PepMV-CH2 isolates are located in the same dark-blue colored cells (arrowhead) of the leaf, mixed together with other noninfected tissue cells, or only infected by one of the isolates. Scale bars are displayed in the images. This figure is adapted with permission from [16].
2.3. Electron Microscopy
Electron microscopy is at the heart of plant virology, and it has been broadly used as the chief method to characterize plant tissues and cells infected by viruses and to describe the alterations induced in infected hosts [32][33][34][35][36][37][38][39][40][41]. Viral particles can be confounded with other particles and/or contaminants in virus preparations or in tissue sections; thus, for specific virus detection, electron microscopy coupled to immunolabeling is the preferred option. Immunogold labelling (IGL), where specific antibodies are linked to gold particles, is a powerful technique for the detection and localization of antigens in thin sections. Gold particles are highly electron dense and thus are able to show the position on a grid of any molecule to which they are attached. IGL has been successfully used for a wide variety of plant virus particles and proteins [42][43][44]. The gold particles are available in a wide range of sizes, from the smaller 1–5 nm ones, to the largest 20–25 nm in size, so choosing antibodies linked to different size particles can allow for detecting more than one antigen in a given tissue section. While IGL has been successfully used in a good number of experimental systems, problems attributable to the fixation and the embedding processes are still a source of recurrent trouble; these steps are crucial for preserving the ultrastructure of the cells as well as immunogenic epitopes. It is thus important, although not always possible, to try different fixation methods, for instance, fixation in glutaraldehyde alone versus glutaraldehyde followed by osmium tetroxide, as well as combinations of glutaraldehyde plus p-formaldehyde. In general terms, glutaraldehyde is better for preserving the cell and tissue structure, while p-formaldehyde provides better antigenic preservation. Similarly, it is important to use appropriate embedding resins such as LR-White or LR-Gold, or low temperature polymerization resins such Lowicryl. Another source of problems may be the specificity of the labelling antibody. In this case, cross absorption of the antiserum with a protein extract from healthy host tissues may result in a significant decrease of the nonspecific signal background.
Specific antibodies can also be used in combination with electron microscopy for the fast and accurate detection of viral particles. Immunosorbent electron microscopy (ISEM) consists of decorating glow discharge-treated TEM grids directly with a coating antibody or with protein A that binds the antibody and immune-immobilized viral particles from a crude tissue sap [45][46][47][48][49]. ISEM seems to be particularly useful in encapsidation/decapsidation studies, where viral particles may be scant and an enrichment process is required prior to observation [50][51].
References
- Más, P.; Pallás, V. Non-isotopic tissue-printing hybridization: A new technique to study long-distance plant virus movement. Virol. Methods 1995, 52, 317–326, doi:10.1016/0166-0934(94)00167-f.
- García-Castillo, S.; Sánchez-Pina, M.A.; Pallás, V.; Kien, F.; Abraham, J.-D.; Schuster, C.; Kieny, M.P. Spatio-temporal analysis of the RNAs, coat and movement (p7) proteins of Carnation mottle virus in Chenopodium quinoa plants. Gen. Virol. 2003, 84, 745–749, doi:10.1099/vir.0.18715-0.
- Marco, C.F.; Aguilar, J.M.; Abad, J.; Gómez-Guillamón, M.L.; Aranda, M.A. Melon Resistance to Cucurbit yellow stunting disorder virus Is Characterized by Reduced Virus Accumulation. Phytopathology 2003, 93, 844–852, doi:10.1094/phyto.2003.93.7.844.
- Gosalvez-Bernal, B.; Garcia-Castillo, S.; Pallás, V.; Sanchez-Pina, M. Distribution of carnation viruses in the shoot tip: Exclusion from the shoot apical meristem. Mol. Plant Pathol. 2006, 69, 43–51, doi:10.1016/j.pmpp.2006.12.004.
- Gosalvez‐Bernal, B.; Genoves, A.; Navarro, J.A.; Pallas, V.; Pina, M.A.S. Distribution and pathway for phloem‐dependent movement of Melon necrotic spot virus in melon plants. Plant Pathol. 2008, 9, 447–461, doi:10.1111/j.1364-3703.2008.00474.x.
- Amari, K.; Burgos, L.; Pallás, V.; Sánchez-Pina, M.A. Vertical transmission of Prunus necrotic ringspot virus: Hitch-hiking from gametes to seedling. Gen. Virol. 2009, 90, 1767–1774, doi:10.1099/vir.0.009647-0.
- Guiu-Aragonés, C.; Sánchez-Pina, M.A.; Díaz-Pendón, J.A.; Pena, E.J.; Heinlein, M.; Martín-Hernández, A.M. cmv1is a gate forCucumber mosaic virus transport from bundle sheath cells to phloem in melon. Plant Pathol. 2016, 17, 973–984, doi:10.1111/mpp.12351.
- Balsalobre, J.M.; Más, P.; Sánchez-Pina, M.A.; Pallás, V. Spatial Distribution of Acidic Chitinases and Their Messenger RNAs in Tobacco Plants Infected with Cherry Leaf Roll Virus. Plant-Microbe Interact. 1997, 10, 784–788, doi:10.1094/mpmi.1997.10.6.784.
- Goldman, V.; Czosnek, H. Whiteflies (Bemisia tabaci) issued from eggs bombarded with infectious DNA clones of Tomato yellow leaf curl virus from Israel (TYLCV) are able to infect tomato plants. Virol. 2002, 147, 787–801, doi:10.1007/s007050200026.
- Gómez, G.; Pallás, V. A Long-Distance Translocatable Phloem Protein from Cucumber Forms a Ribonucleoprotein Complex In Vivo with Hop Stunt Viroid RNA. Virol. 2004, 78, 10104–10110, doi:10.1128/jvi.78.18.10104-10110.2004.
- Peter, K.A.; Gildow, F.; Palukaitis, P.; Gray, S.M. The C Terminus of the Polerovirus P5 Readthrough Domain Limits Virus Infection to the Phloem. Virol. 2009, 83, 5419–5429, doi:10.1128/jvi.02312-08.
- Collum, T.D.; Culver, J.N. Tobacco mosaic virus infection disproportionately impacts phloem associated translatomes in Arabidopsis thaliana and Nicotiana benthamiana. Virology 2017, 510, 76–89, doi:10.1016/j.virol.2017.07.002.
- Garcı́a-Castillo S.; Marcos, J.F.; Pallas, V.; Pina, M.A.S. Influence of the plant growing conditions on the translocation routes and systemic infection of carnation mottle virus in Chenopodium quinoa plants. Mol. Plant Pathol. 2001, 58, 229–238, doi:10.1006/pmpp.2001.0332.
- Amari, K.; Burgos, L.; Pallas, V.; Sanchez-Pina, M.A. Prunus necrotic ringspot virus Early Invasion and Its Effects on Apricot Pollen Grain Performance. Phytopathology 2007, 97, 892–899, doi:10.1094/phyto-97-8-0892.
- Rajamäki, M.-L.; Valkonen, J.P.T. Viral Genome-Linked Protein (VPg) Controls Accumulation and Phloem-Loading of a Potyvirus in Inoculated Potato Leaves. Plant-Microbe Interact. 2002, 15, 138–149, doi:10.1094/mpmi.2002.15.2.138.
- Gómez-Aix, C.; Alcaide, C.; Gómez, P.; Aranda, M.; Sánchez-Pina, M. In situ hybridization for the localization of two pepino mosaic virus isolates in mixed infections. Virol. Methods 2019, 267, 42–47, doi:10.1016/j.jviromet.2019.02.003.
- Wan, J.; Basu, K.; Mui, J.; Vali, H.; Zheng, H.; Laliberté, J.-F. Ultrastructural Characterization of Turnip Mosaic Virus-Induced Cellular Rearrangements Reveals Membrane-Bound Viral Particles Accumulating in Vacuoles. Virol. 2015, 89, 12441–12456, doi:10.1128/jvi.02138-15.
- Aranda, M.A.; Escaler, M.; Wang, D.; Maule, A.J. Induction of HSP70 and polyubiquitin expression associated with plant virus replication. Natl. Acad. Sci. USA 1996, 93, 15289–15293, doi:10.1073/pnas.93.26.15289.
- Aranda, M.A.; Maule, A. Virus-Induced Host Gene Shutoff in Animals and Plants. Virology 1998, 243, 261–267, doi:10.1006/viro.1998.9032.
- Amari, K.; Díaz-Vivancos, P.; Pallás, V.; Sánchez-Pina, M.A.; Hernández, J.A. Oxidative stress induction by Prunus necrotic ringspot virus infection in apricot seeds. Plant. 2007, 131, 302–310, doi:10.1111/j.1399-3054.2007.00961.x.
- Jackson, D. In situ hybridization in plants. In Molecular Plant Pathology: A Practical Approach; Gurr, S.J., McPherson, M.J., Eds.; IRL Press: Oxford, UK, 1991; pp. 163–174.
- Lim, H.-S.; Vaira, A.M.; Bae, H.; Bragg, J.N.; Ruzin, S.E.; Bauchan, G.R.; Dienelt, M.M.; Owens, R.A.; Hammond, J. Mutation of a chloroplast-targeting signal in Alternanthera mosaic virus TGB3 impairs cell-to-cell movement and eliminates long-distance virus movement. Gen. Virol. 2010, 91, 2102–2115, doi:10.1099/vir.0.019448-0.
- Sicard, A.; Pirolles, E.; Gallet, R.; Vernerey, M.-S.; Yvon, M.; Urbino, C.; Peterschmitt, M.; Gutierrez, S.; Michalakis, Y.; Blanc, S. A multicellular way of life for a multipartite virus. eLife 2019, 8, 1–14, doi:10.7554/elife.43599.
- Silva, G.; Souza, M. Review Genomic in situ hybridization in plants. Mol. Res. 2013, 12, 2953–2965, doi:10.4238/2013.august.12.11.
- Johansen, B. In situPCR on Plant Material with Sub-cellular Resolution. Bot. 1997, 80, 697–700, doi:10.1006/anbo.1997.0502.
- Rojas, M.R.; Jiang, H.; Salati, R.; Xoconostle-Cázares, B.; Sudarshana, M.; Lucas, W.J.; Gilbertson, R.L. Functional Analysis of Proteins Involved in Movement of the Monopartite Begomovirus, Tomato Yellow Leaf Curl Virus. Virology 2001, 291, 110–125, doi:10.1006/viro.2001.1194.
- Silva, C.; Tereso, S.; Nolasco, G.; Oliveira, M.M. Cellular Location of Prune dwarf virus in Almond Sections by In Situ Reverse Transcription-Polymerase Chain Reaction. Phytopathology 2003, 93, 278–285, doi:10.1094/phyto.2003.93.3.278.
- Pesquet, E.; Barbier, O.; Ranocha, P.; Jauneau, A.; Goffner, D. Multiple gene detection byin situRT-PCR in isolated plant cells and tissues. Plant J. 2004, 39, 947–959, doi:10.1111/j.1365-313x.2004.02170.x.
- Yoo, B.-C.; Kragler, F.; Varkonyi-Gasic, E.; Haywood, V.; Archer-Evans, S.; Lee, Y.M.; Lough, T.J.; Lucas, W.J. A Systemic Small RNA Signaling System in Plants. Plant Cell 2004, 16, 1979–2000, doi:10.1105/tpc.104.023614.
- Ham, B.-K.; Brandom, J.L.; Xoconostle-Cázares, B.; Ringgold, V.; Lough, T.J.; Lucas, W.J. A Polypyrimidine Tract Binding Protein, Pumpkin RBP50, Forms the Basis of a Phloem-Mobile Ribonucleoprotein Complex. Plant Cell 2009, 21, 197–215, doi:10.1105/tpc.108.061317.
- Chang, C.-H.; Hsu, F.-C.; Lee, S.-C.; Lo, Y.-S.; Wang, J.-D.; Shaw, J.; Taliansky, M.; Chang, B.-Y.; Hsu, Y.-H.; Lin, N.-S. The Nucleolar Fibrillarin Protein Is Required for Helper Virus-Independent Long-Distance Trafficking of a Subviral Satellite RNA in Plants. Plant Cell 2016, 28, 2586–2602, doi:10.1105/tpc.16.00071.
- Carroll, T.W.; Mayhew, D.E. Anther and pollen infection in relation to the pollen and seed transmissibility of two strains of barley stripe mosaic virus in barley. J. Bot. 1976, 54, 1604–1621, doi:10.1139/b76-173.
- Carroll, T.W.; Mayhew, D.E. Occurrence of virions in developing ovules and embryo sacs of barley in relation to the seed transmissibility of barley stripe mosaic virus. J. Bot. 1976, 54, 2497–2512, doi:10.1139/b76-268.
- Hunter, D.G.; Bowyer, J.W. Cytopathology of Developing Anthers and Pollen Mother Cells from Lettuce Plants Infected by Lettuce Mosaic Potyvirus. Phytopathol. 1997, 145, 521–524, doi:10.1111/j.1439-0434.1997.tb00360.x.
- Roberts, I.M.; Wang, D.; Thomas, C.L.; Maule, A.J. Pea seed-borne mosaic virus seed transmission exploits novel symplastic pathways to infect the pea embryo and is, in part, dependent upon chance. Protoplasma 2003, 222, 31–43, doi:10.1007/s00709-003-0015-5.
- Minicka, J.; Otulak, K.; Garbaczewska, G.; Pospieszny, H.; Hasiów-Jaroszewska, B. Ultrastructural insights into tomato infections caused by three different pathotypes of Pepino mosaic virus and immunolocalization of viral coat proteins. Micron 2015, 79, 84–92, doi:10.1016/j.micron.2015.08.006.
- Sempere, R.N.; Gómez-Aix, C.; Ruíz-Ramón, F.; Gómez, P.; Hasiów-Jaroszewska, B.; Sánchez-Pina, M.A.; Aranda, M.A. Pepino mosaic virus RNA-Dependent RNA Polymerase POL Domain Is a Hypersensitive Response-Like Elicitor Shared by Necrotic and Mild Isolates. Phytopathology 2016, 106, 395–406, doi:10.1094/phyto-10-15-0277-r.
- Tilsner, J.; Linnik, O.; Christensen, N.M.; Bell, K.; Roberts, I.M.; Lacomme, C.; Oparka, K.J. Live-cell imaging of viral RNA genomes using a Pumilio-based reporter. Plant J. 2009, 57, 758–770, doi:10.1111/j.1365-313x.2008.03720.x.
- Tilsner, J.; Linnik, O.; Wright, K.M.; Bell, K.; Roberts, A.G.; Lacomme, C.; Cruz, S.S.; Oparka, K.J. The TGB1 Movement Protein of Potato virus X Reorganizes Actin and Endomembranes into the X-Body, a Viral Replication Factory. Plant Physiol. 2012, 158, 1359–1370, doi:10.1104/pp.111.189605.
- Tilsner, J.; Linnik, O.; Louveaux, M.; Roberts, I.M.; Chapman, S.N.; Oparka, K.J. Replication and trafficking of a plant virus are coupled at the entrances of plasmodesmata. Cell Biol. 2013, 201, 981–995, doi:10.1083/jcb.201304003.
- Zechmann, B.; Zellnig, G. Cytological modifications in zucchini yellow mosaic virus (ZYMV)-infected Styrian pumpkin plants. Virol. 2003, 148, 1119–1133, doi:10.1007/s00705-003-0005-0.
- Brandizzi, F.; Frangne, N.; Marc-Martin, S.; Hawes, C.; Neuhaus, J.-M.; Paris, N. The Destination for Single-Pass Membrane Proteins Is Influenced Markedly by the Length of the Hydrophobic Domain. Plant Cell 2002, 14, 1077–1092, doi:10.1105/tpc.000620.
- Ju, H.-J.; Samuels, T.D.; Wang, Y.-S.; Blancaflor, E.; Payton, M.; Mitra, R.; Krishnamurthy, K.; Nelson, R.S.; Verchot-Lubicz, J. The Potato Virus X TGBp2 Movement Protein Associates with Endoplasmic Reticulum-Derived Vesicles during Virus Infection1. Plant Physiol. 2005, 138, 1877–1895, doi:10.1104/pp.105.066019.
- Wan, J.; Cabanillas, D.G.; Zheng, H.; Laliberté, J.-F. Turnip mosaic virus Moves Systemically through Both Phloem and Xylem as Membrane-Associated Complexes. Plant Physiol. 2015, 167, 1374–1388, doi:10.1104/pp.15.00097.
- Milne, R.; Lesemann, D.-E. Immunosorbent Electron Microscopy in Plant Virus Studies. Methods Virol. 1984, VIII, 85–101, doi:10.1016/b978-0-12-470208-0.50009-8.
- Ritzenthaler, C.; Laporte, C.; Gaire, F.; Dunoyer, P.; Schmitt, C.; Duval, S.; PiéquetA.; Loudes, A.M.; Rohfritsch, O.; Stussi-Garaud, C.; et al. Grapevine Fanleaf Virus Replication Occurs on Endoplasmic Reticulum-Derived Membranes. Virol. 2002, 76, 8808–8819, doi:10.1128/jvi.76.17.8808-8819.2002.
- Kaplan, I.B.; Lee, L.; Ripoll, D.R.; Palukaitis, P.; Gildow, F.; Gray, S.M. Point mutations in the potato leafroll virus major capsid protein alter virion stability and aphid transmission. Gen. Virol. 2007, 88, 1821–1830, doi:10.1099/vir.0.82837-0.
- Smirnova, E.; Firth, A.E.; Miller, W.A.; Scheidecker, D.; Brault, V.; Reinbold, C.; Rakotondrafara, A.M.; Chung, B.Y.-W.; Ziegler-Graff, V. Discovery of a Small Non-AUG-Initiated ORF in Poleroviruses and Luteoviruses That Is Required for Long-Distance Movement. PLoS Pathog. 2015, 11, e1004868, doi:10.1371/journal.ppat.1004868.
- Laue, M. Electron Microscopy of Viruses; Elsevier BV: Amsterdam, The Netherlands, 2010; Volume 96, pp. 1–20.
- Lough, T.J.; Netzler, N.E.; Emerson, S.J.; Sutherland, P.; Carr, F.; Beck, D.L.; Lucas, W.J.; Forster, R.L.S. Cell-to-Cell Movement of Potexviruses: Evidence for a Ribonucleoprotein Complex Involving the Coat Protein and First Triple Gene Block Protein. Plant-Microbe Interact. 2000, 13, 962–974, doi:10.1094/mpmi.2000.13.9.962.
- De, S.; Pollari, M.; Varjosalo, M.; Mäkinen, K. Association of host protein VARICOSE with HCPro within a multiprotein complex is crucial for RNA silencing suppression, translation, encapsidation and systemic spread of potato virus A infection. PLoS Pathog. 2020, 16, e1008956, doi:10.1371/journal.ppat.1008956.

