 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Szymon Hryhorowicz | + 3109 word(s) | 3109 | 2021-02-26 06:53:14 | | | |
| 2 | Peter Tang | Meta information modification | 3109 | 2021-03-06 16:13:43 | | |
Video Upload Options
The endocannabinoid system (ECS) is an endogenous signaling system formed by specific receptors (cannabinoid type 1 and type 2 (CB1 and CB2)), their endogenous ligands (endocannabinoids), and enzymes involved in their synthesis and degradation. The ECS, centrally and peripherally, is involved in various physiological processes, including regulation of energy balance, promotion of metabolic process, food intake, weight gain, promotion of fat accumulation in adipocytes, and regulation of body homeostasis; thus, its overactivity may be related to obesity.
1. Introduction
According to the World Health Organization (WHO), obesity is defined by a body mass index (BMI) higher than or equal to 30 kg/m2. Although the BMI is not an ideal diagnostic tool, it is widely used in clinical practice as well as in assessing the prevalence of obesity worldwide. However, it should be noted that the BMI uses only height and body weight, which could be misleading when muscle tissue is overgrown [1]. Therefore, the waist-to-hip ratio (WHR) should be measured to assess the location of adipose tissue [2]. In recent years, more accurate techniques, such as bioelectrical impedance analysis (BIA) or gold-standard dual-energy X-ray absorptiometry (DXA), have become more common, allowing a more precise evaluation of adipose tissue [3].
Within a few decades, obesity has become a global problem. Currently, almost 2 billion individuals worldwide suffer from overweight, and over 650 million are obese (Figure 1) [4]. In Poland, 21.3% of adults suffered from obesity in 2016, with a slight predominance of men. On the other hand, in the U.S., 36.2% of the population, predominantly women, suffer from obesity [5]. In fact, the prevalence of obesity has almost tripled since 1975 [6], and it is estimated that by 2030, half of the world’s population will be overweight [7]. Currently, obesity, especially among children, is vastly increasing in developing countries, particularly in urban areas. Interestingly, the prevalence of obesity is more than 30% higher among children living in developing countries than among children living in developed countries [8]. It is essential to note that 75% of people with excessive body weight in childhood will suffer from overweight or obesity in adult life [9]. The pathogenesis of obesity is complex and has not been investigated thoroughly enough, although it is accepted that behavioral, genetic, and biological factors, including intestinal microbiota or even intrauterine growth, have been associated with the development of obesity [8][10][11].
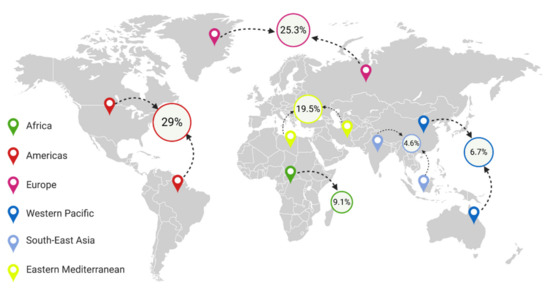
Figure 1. Percentage of people with obesity in different regions of the world [12][13][14][15][16].
There is evidence of a link between intestinal microbiota and obesity. It has been discovered that microbiological changes in the intestine constitute a risk factor for obesity among humans [17]. In addition, bariatric surgery partly improves excessive body weight, associated with intestinal dysbiosis, and the changes in the intestinal microbiota composition have been associated with positive results following the surgery, i.e., with weight loss or metabolism improvement [11].
However, the direct cause of obesity is excessive calorie intake. Nearly 3 million people in the world die of obesity-related comorbidities each year [5]. Furthermore, obesity is associated with a higher incidence of cardiovascular disease, including heart failure, which is the main cause of death globally.
One of the elements responsible for the body’s nutrition and metabolic state is the food-intake-regulating system, involving numerous hormones, cytokines, and other transmitters, such as insulin, leptin, ghrelin, glucocorticosteroids, and endocannabinoids [18]. Food intake is regulated at both central and peripheral levels [19]. Central regulation is responsible for nutritional (pro-nutritional) behavior and the resulting feeling of pleasure, whereas peripheral regulation is based on the modification of lipogenesis, adipogenesis, glucose metabolism, and lipoprotein metabolism, in which the endocannabinoid system (ECS) plays a significant role [4][20].
The isolation of Δ9-tetrahydrocannabinol (Δ9-THC) in 1964 was a milestone in discovering the endocannabinoid system [21]. Studies on Δ9-THC led to the identification of the first cannabinoid receptor, i.e., cannabinoid receptor type 1 (CB1R) in the rat brain, which was classified as a G-protein-coupled receptor (GPCR) [22]. In the following years, CB1R was found in many species, including humans [23]. In 1993, a second cannabinoid receptor, cannabinoid receptor type 2 (CB2R), also classified as a GPCR, was discovered in spleen macrophages [24]. It is worth mentioning that other types of cannabinoid receptors, nonCB1 and nonCB2 orphan GPR55 and TRPV1 receptors, have been reported so far, which may explain the pathway that has not been fully understood [25][26][27]. However, to date, no genetic polymorphism changes in GPR55 and TRPV receptors have been identified in terms of their role in determining obesity. Only in vivo studies on the role of GPR55 in energy and glucose homeostasis have been performed in GPR55-/- mice, which revealed that GPR55 knockout, at least partially, increased adiposity and insulin resistance due to reduced physical activity [28]. A similar observation was confirmed for the TRPV receptor, where the authors evaluated the effect of capsaicin on the browning program in white adipose tissue (WAT) by the activation of TRPV1 channels to prevent diet-induced obesity in wild-type and TRPV1(-/-) mouse models. They successfully demonstrated that activation of TRPV1 channels by dietary capsaicin results in the browning of WAT, thus prevent obesity, which implies that TRPV could become a promising new target to combat obesity [29]. This fact was confirmed by Christie et al. in 2018, which indicates that TRPV may be involved in energy homeostasis and the control of food intake, appetite, and energy expenditure. This, in turn, strongly suggests that its dysregulation may be involved in the development of obesity. The mechanisms causing dysregulation have not been fully understood, but interactions with the ECS may, to some extent, explain the role of TRPV in this dysregulation [30].
The discovery of cannabinoid receptors allowed one to identify its endogenous cannabinoids, such as the endogenous partial agonist anandamide (AEA) and the endogenous full agonist 2-arachidonylglycerol (2-AG) [31], with a greater affinity for CB1 and CB2 than AEA [32][33], or virodhamine (CB1 receptor antagonist and CB2 receptor agonist) derived from arachidonic acid and ethanolamine [34].
The abovementioned AEA and 2-AG are the best-known endocannabinoids, although other ECS neurotransmitters, such as 2-arachidonyl glyceryl ether (2-AGE), N-arachidonoyl dopamine (NADA), oleamide (cis-9,10-octadecanoamide (ODA)), N-arachidonylglycine (NAGLy), palmitoylethanolamide (PEA), stearoylethanolamide (SEA), and oleoylethanolamine (OEA) [33], should also be mentioned, since they all have an affinity for cannabinoid-like G-coupled receptors. The endocannabinoid system is also formed by synthesizing and degrading enzymes, such as AEA-synthesizing enzymes (N- acylotransferase (NAT), N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD), and fatty acid amide hydrolase (FAAH)) and 2-AG-regulating enzymes (diacylglycerol lipase (DAGL) and monoacylglycerol lipase (MAGL)) [33][35]. Moreover, it has been recently discovered that glycerophosphodiester phosphodiesterase 3 (GDE3) acts as an ecto-enzyme and converts bioactive lysophosphadylinositol (LPI) to monoacyloglycerols (MG), including 2-AG, and activates CB1R as well as CB2R signaling in mammalian cultured cells [36][37].
CB1R is primarily located in the central and peripheral nervous systems, i.e., in the cerebral cortex (neocortex), with a high accumulation in the cingulate cortex, and the frontal and motor cortex. It is also present in the olfactory structures of the brain and the hippocampus, where the accumulation is exceptionally high, as well as in the amygdala, striatum, cortex, deep cerebellar nuclei, brain stem, spinal cord, diencephalon, and hypothalamus, where, in contrast, its accumulation is relatively low [38]. It should be noted that CB1R is the most common receptor of GPCRs in the mammalian central nervous system (CNS) [39][40]. Besides, CB1R is found in adipocytes and muscles, adrenals, pancreas, liver, gastrointestinal cells, and other tissues [41]. CB2R is formerly considered a peripheral receptor due to its location in spleen macrophages. However, cannabinoid receptor type 2 has also been found in some parts of the brain, such as the striatum, hypothalamus, cerebral cortex, hippocampus, amygdala, and substantia nigra [41][42]. Nevertheless, the immune system is the primary location of the CB2 receptor. The CB2 receptor was found in macrophages, including osteocytes, osteoclasts, Kupffer cells, and B lymphocytes, and in each organ with immune cells, including the cardiovascular system, gastrointestinal tract, and reproductive system, and plays an important role in inflammatory processes (Table 1) [43][44][45][46].
Table 1. Presence of CB1R and CB2R in human tissues [47][48][49].
|
Concentration Tissue |
CB1R |
CB2R |
|---|---|---|
|
Adrenals |
+ + |
0 |
|
Appendix |
+ + |
+ + |
|
Bone marrow |
Low |
+ |
|
Brain |
+ + + + |
0 |
|
Colon |
+ |
Low |
|
Duodenum |
+ |
Low |
|
Endometrium |
+ |
Very low |
|
Esophagus |
+ |
Very low |
|
Fat |
+ + + |
0 |
|
Gall bladder |
+ |
|
|
Heart |
Low |
0 |
|
Kidney |
Very low |
0 |
|
Liver |
0 |
Very low |
|
Lung |
+ + |
Low |
|
Lymph node |
+ + |
+ + + |
|
Ovary |
+ |
0 |
|
Pancreas |
Very low |
0 |
|
Placenta |
+ + |
Very low |
|
Prostate gland |
+ |
Low |
|
Salivary gland |
+ |
Very low |
|
Skin |
+ |
Very low |
|
Small intestine |
+ |
Low |
|
Spleen |
+ |
+ + + |
|
Stomach |
+ |
+ |
|
Testis |
+ |
Low |
|
Thyroid |
+ |
Low |
|
Urinary bladder |
+ |
+ |
RNA-sequencing. RPKM, reads per kilobase million. 0, absent; very low, concentrations equal and below 0.03 RPKM; low, concentrations in the range of 0.119–0.031 RPKM; +, concentrations in the range of 0.582–0.200 RPKM; + +, concentrations in the range of 1.272–1.089 RPKM; + + +, concentrations in the range of 3.665–2.735 RPKM; + + + +, concentration equal to 6.155 RPKM; CB1R—cannabinoid receptor type 1; CB2R—cannabinoid receptor type 2.
2. Role of the Endocannabinoid System in Metabolic Process Regulation
Endocannabinoids are involved in the physiological regulation of the body’s homeostasis, stimulating food intake and hunger, as well as shifting energy balance toward energy storage [50], by means of acting on peripheral tissues, such as adipocytes, hepatocytes, islet cells, the gastrointestinal tract, and skeletal muscles (Figure 2).
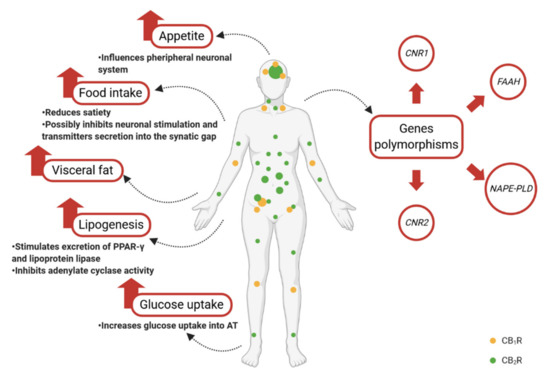
Figure 2. Schematic illustration of the endocannabinoid system role in the regulation of metabolic processes. CB1R—cannabinoid receptor type 1; CB2R—cannabinoid receptor type 2; CNR1—endocannabinoid type 1 receptor gene; CNR2—endocannabinoid type 2 receptor gene; FAAH—fatty acid amide hydrolase; NAPE-PLD—N-acyl phosphatidylethanolamine phospholipase D.
2.1. Adipose Tissue
The ECS promotes fat storage in adipocytes by intensifying adipogenesis and acts directly by increasing triacylglyceride (TAG) production. It has been shown that the blockage of CB1R decreases adipocyte proliferation, while adipocyte differentiation is directly preceded by multiple increases in AEA and 2-AG levels in mice [51][52]. CB1R stimulation is accompanied by an increase in peroxisome proliferator-activated receptor gamma (PPAR-γ) receptors, which play an essential role in adipocyte proliferation and increase the size and quantity of TAG in adipocytes of diet-induced obese mice [51][53]. Additionally, CB1R activation decreases adiponectin expression and increases leptin expression in mouse white adipose tissue (WAT) [54].
2.2. Lipogenesis and Lipolysis
CB1R activation in mice stimulates the expression of PPAR-γ and lipoprotein lipase, which increases the availability of substrate for TAG production, stored in adipocytes [55]. Moreover, CB1 inhibits adenylate cyclase activity, which inhibits the activity of 5′AMP-activated protein kinase (AMPK), and further of AMPK-associated lipolysis, which reduces fatty acid synthase (FAS) inhibition and lipolysis. CB1R stimulation also directly increases FAS expression. On the other hand, CB1R increases glucose uptake into the adipose tissue by directly affecting glucose transporter type 4 (GLUT4) in adipocytes. These activities under physiological conditions are regulated by autocrine mechanisms in the adipose tissue. However, under pathological conditions, these actions cause dyslipidemia, i.e., mainly an increase in TAG and low-density lipoprotein (LDL) in the serum, and play a crucial role in developing insulin resistance (IR) [20].
2.3. Brown Adipose Tissue
CB1R is present in brown adipose tissue (BAT) adipocytes. Previous studies suggest that CB1R activation in BAT is based on the inhibition of signals from the sympathetic nervous system (SNS), which decreases thermogenesis [56]. Moreover, peripheral CB1R blockade in BAT can provide a new approach to treating obesity and lowering cardiovascular risk. In fact, chronic CB1R antagonism has been associated with activation of BAT thermogenesis and weight loss in diet-induced obese mice and rats by both peripheral and CNS-located CB1R [56][57]. As Eriksson et al. demonstrated, CB1R can be a promising surrogate biomarker for BAT, which would be helpful in further investigation of the activation and regulation of BAT and energy expenditure [58]. Similar results were obtained in the study by Boon et al., where CB1R blockade with rimonabant in mice enhanced energy expenditure and reduced dyslipidemia [59]. However, EC signaling via CB1R could also provide new approaches to treating obesity and improving metabolism in humans [60].
2.4. Liver
As mentioned earlier, CB1R is expressed in the liver, promoting the synthesis and storage of TAG [61]. The activation of CB1R in wild-type mice resulted in increased expression of sterol regulatory element-binding protein-1c (SREBP-1c) and subsequent expression of related lipogenic enzymes, e.g., FAS and acetyl-CoA carboxylase. Under pathological conditions, it significantly contributes to the development of non-alcoholic steatohepatitis (NASH), further hepatic fibrosis, and dyslipidemia or dyslipoproteinemia [62][63][64]. Moreover, CB1R activation causes an expansion of the adipose tissue in the liver, which causes IR [65]. In contrast, the ECS inhibits adiponectin expression, which stimulates liver AMPK and fatty acid entrance into mitochondrial oxidation pathways. It proves that in addition to the direct effect on liver metabolism, the ECS also indirectly decreases the oxidation of fatty acids in the liver in vivo [66]. Furthermore, CB2 is also expressed in hepatocytes, and CB1R antagonists and CB2 agonists protect the liver from toxic failure [67].
2.5. Skeletal Muscles
The action effect of CB1 in the muscles is associated with the regulation of glucose uptake through the modulation of insulin sensitivity. Under physiological conditions, the activation of CB1 in mice results in a decrease in glucose uptake and oxygen consumption by inhibiting fatty acid oxidation [68][69].
2.6. Pancreas
It has been demonstrated that CB1R and CB2 are present in alpha and beta islet cells, although their stimulation has a different result. The activation of CB1 increases insulin secretion from beta cells and glucagon from alpha cells in mice. CB2 stimulation decreases insulin secretion from beta cells and glucagon from alpha cells. These data suggest, therefore, that the glucose plasma concentrations induced by the ECS are not directly associated with pancreatic hormones; however, they are associated to a greater extent with the modulation of peripheral insulin sensitivity dependent upon the ECS [20][70][71].
2.7. Gastrointestinal Tract
The ECS reduces the feeling of satiety, which increases the frequency and quantity of food intake. Simultaneously, the ECS decreases gastric juice secretion, intestinal peristalsis, and bowel content passage. This, in turn, results in a higher absorption of nutrients, which could lead to weight gain and further obesity. These processes are influenced by receptors located in the gastrointestinal tract (GT) and the peripheral nervous system [72][73][74]. In fact, a high-fat diet-specific increase of AEA and 2-AG in the jejunum suggests the presence of positive feedback loops. The inhibition of this loop could reduce the consumption of fat-rich products, thus providing a therapeutic solution [75][76]. Furthermore, endocannabinoids are also produced in the gastrointestinal tract, e.g., in the small intestine. On the basis of the mice model, it has been indicated that after 24 h fasting, the level of AEA in the GT increases sevenfold, which can affect the nutritional status, energy balance, lipoprotein metabolism, glucose homeostasis, and even nutritional behavior [39][72].
3. Cannabinoids/Endocannabinoid Control of Food Intake
ECS activity in the central nervous system—mostly in the limbic system and hypothalamus—is well documented. The endocannabinoid system plays an essential role in connecting gastrointestinal tract activity and the body’s energy economy [50]. The ECS works by increasing both appetite and the motivation to seek food. This mechanism is regulated by nourishment and anorexigenic transmitters produced by the hypothalamus, e.g., corticotropin-releasing hormone (CRH), melanin-concentrating hormone (MCH), and hypocretin [77][78].
It is assumed that the ECS operates at the cellular level by inhibiting neuronal stimulation and transmitters secretion into the synaptic gap [79]. The EC level in the hypothalamus is physiologically regulated by hormones reflecting the organism’s metabolic conditions, e.g., leptin, ghrelin, cholecystokinin, and glucocorticosteroids. The use of cannabinoids increases food intake [80]. Studies on rodents have indicated that those with 60% deficiency of CB1R in the hypothalamus are less sensitive to a non-nutritive effect of rimonabant (a selective agonist of CB1R, previously used as an anti-obesity treatment and then excluded due to multiple adverse reactions, mostly psychiatric). Interestingly, it has been suggested that the leptin effect in the hypothalamus is mostly associated with ECS signaling, since no leptin-induced appetite suppression effect was observed in rodents. Moreover, in another study, ghrelin also reduced this effect [54][81].
It has been suggested that several neuronal connections, directly related to eating behaviors, change their function from stimulation to obesity inhibition. Therefore, EC’s presynaptic inhibiting effect on the expression of transmitters stimulates the expression of pro-nourishing transmitters [54]. Considering that leptin negatively regulates the expression of CB1R in the hypothalamus and the fact that leptin resistance is common, leptin resistance impairs negative leptin control of the ECS at the hypothalamus level, causing an increase in subsequent pro-nourishing behaviors [4][82][83]. Moreover, the administration of the reverse CB1R agonist restores leptin sensitivity and has an anti-obesity effect in mice [84]. Other rodent studies revealed an increase of 2-AG in the hypothalamus following a high-fat diet (regardless of chronic or acute stimulation). The selective activation of CB1R in the central nervous system resulted in resistance to obesity induced by a high-fat diet. Moreover, the inactivation of CB1R in the central and sympathetic nervous systems caused an increase of thermogenesis [54][74][82][85][86].
The role of the limbic system in the control of food intake is mainly based on hedonistic fulfilment and needs—in this case, the estimation of taste and related behaviors. The nucleus accumbens, where endocannabinoid and opioid receptors are found, plays the most crucial role in assessing food type. Additionally, the nucleus accumbens has a unique link with the lateral hypothalamus, where the ECS performs a significant function [87][88]. In fact, it has been proven that dopamine activity in the nucleus accumbens is associated with the classification of hedonistic impulses and that CB1R blockage in this structure inhibits dopamine expression in response to pleasurable food [87][89]. Furthermore, the vagus nerve contains CB1R, CCK, and leptin receptors and is also responsible for maintaining homeostasis. It transfers information from organs to the locus coeruleus concerned with the regulation of digestive processes. CCK is secreted from the duodenum during eating and reduces food intake via the vagus nerve. In addition, leptin negatively regulates CB1R levels, also in vagus nerve endings, which suggests another theoretical ECS-related mechanism of reducing food consumption [82].
Moreover, chronic activation of CB1R may increase processes associated with hyperlipidemia, diabetes, or cardiovascular events among hedonic patients with obesity [20][90]. Furthermore, several dietary factors, e.g., dietary secondary metabolites, can affect the ECS, and high-calorie and high-fat diets can modulate the CB1/CB2 ratio and, thus, enhance food intake in several cases [91][92]. EC can also moderate food reward, whereas EC agonists can increase the palatability and hedonic value of food [93]. Interestingly, there is a possible 6-n-propylthiouracil (PROP) taste sensitivity association with the BMI, lipid parameters, and circulating endocannabinoids. Therefore, lower concentrations of AEA or 2-AG in normal-weight nontasters versus normal-weight supertasters can counteract the excess adipose tissue accumulation. In contrast, obesity can disrupt the abovementioned adaptive mechanism, since Carta et al. also noted an opposite correlation between plasma AEA and 2-AG concentrations, as well as the PROP phenotype. As a result, nontasters had 62% higher levels of endocannabinoids than supertasters [94]. It is worth mentioning that the ECS is also dysregulated in eating disorders, and EC dysregulation can be a modulating factor of rewarding binge-eating or self-starvation; however, data regarding this issue remains limited [95].
References
- Batsis, J.A.; Villareal, D.T. Sarcopenic obesity in older adults: Aetiology, epidemiology and treatment strategies. Nat. Rev. Endocrinol. 2018, 14, 513–537.
- Neeland, I.J.; Poirier, P.; Després, J.-P. Cardiovascular and metabolic heterogeneity of obesity: Clinical challenges and implications for management. Circulation 2018, 137, 1391–1406.
- Martin-Calvo, N.; Moreno-Galarraga, L.; Martinez-Gonzalez, M.A. Association between body mass index, waist-to-height ratio and adiposity in children: A systematic review and meta-analysis. Nutrients 2016, 8, 512.
- Rossi, F.; Punzo, F.; Umano, G.R.; Argenziano, M.; Miraglia Del Giudice, E. Role of cannabinoids in obesity. Int. J. Mol. Sci. 2018, 19, 2690.
- WHO (World Health Organization). Available online: https://www.who.int/ (accessed on 2 August 2020).
- Schetz, M.; De Jong, A.; Deane, A.M.; Druml, W.; Hemelaar, P.; Pelosi, P.; Pickkers, P.; Reintam-Blaser, A.; Roberts, J.; Sakr, Y.; et al. Obesity in the critically ill: A narrative review. Intensive Care Med. 2019, 45, 757–769.
- Finkelstein, E.A.; Khavjou, O.A.; Thompson, H.; Trogdon, J.G.; Pan, L.; Sherry, B.; Dietz, W. Obesity and severe obesity forecasts through. Am. J. Prev. Med. 2012, 42, 563–570.
- Di Cesare, M.; Sorić, M.; Bovet, P.; Miranda, J.J.; Bhutta, Z.; Stevens, G.A.; Laxmaiah, A.; Kengne, A.-P.; Bentham, J. The epidemiological burden of obesity in childhood: A worldwide epidemic requiring urgent action. BMC Med. 2019, 17.
- Freedman, D.S.; Khan, L.K.; Dietz, W.H.; Srinivasan, S.R.; Berenson, G.S. Relationship of childhood obesity to coronary heart disease risk factors in adulthood: The bogalusa heart study. Pediatrics 2001, 108, 712–718.
- Taveras, E.M.; Rifas-Shiman, S.L.; Belfort, M.B.; Kleinman, K.P.; Oken, E.; Gillman, M.W. Weight status in the first 6 months of life and obesity at 3 years of age. Pediatrics 2009, 123, 1177–1183.
- Debédat, J.; Clément, K.; Aron-Wisnewsky, J. Gut microbiota dysbiosis in human obesity: Impact of bariatric surgery. Curr. Obes. Rep. 2019, 8, 229–242.
- Prevalence of Obesity among Adults, BMI >= 30 (Crude Estimate) (%). Available online: https://www.who.int/data/maternal-newborn-child-adolescent/monitor (accessed on 2 August 2020).
- Smith, K.B.; Smith, M.S. Obesity statistics. Prim. Care 2016, 43, 121–135.
- Chooi, Y.C.; Ding, C.; Magkos, F. The epidemiology of obesity. Metab. Clin. Exp. 2019, 92, 6–10.
- Yumuk, V.; Tsigos, C.; Fried, M.; Schindler, K.; Busetto, L.; Micic, D.; Toplak, H. Obesity management task force of the European Association for the study of obesity European guidelines for obesity management in adults. Obes. Facts 2015, 8, 402–424.
- Misra, A.; Jayawardena, R.; Anoop, S. Obesity in South Asia: Phenotype, morbidities, and mitigation. Curr. Obes. Rep. 2019, 8, 43–52.
- Abenavoli, L.; Scarpellini, E.; Colica, C.; Boccuto, L.; Salehi, B.; Sharifi-Rad, J.; Aiello, V.; Romano, B.; De Lorenzo, A.; Izzo, A.A.; et al. Gut microbiota and obesity: A role for probiotics. Nutrients 2019, 11, 2690.
- Ahima, R.S.; Antwi, D.A. Brain regulation of appetite and satiety. Endocrinol. Metab. Clin. North Am. 2008, 37, 811–823.
- Lenard, N.R.; Berthoud, H.-R. Central and peripheral regulation of food intake and physical activity: Pathways and genes. Obesity (Silver Spring) 2008, 16, S11–S22.
- Di Marzo, V. The endocannabinoid system in obesity and type 2 diabetes. Diabetologia 2008, 51, 1356.
- Crocq, M.-A. History of cannabis and the endocannabinoid system. Dialogues Clin. Neurosci 2020, 22, 223–228.
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned CDNA. Nature 1990, 346, 561–564.
- Gérard, C.M.; Mollereau, C.; Vassart, G.; Parmentier, M. Molecular cloning of a human cannabinoid receptor which is also expressed in testis. Biochem. J. 1991, 279, 129–134.
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65.
- Bisogno, T.; Hanus, L.; De Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharm. 2001, 134, 845–852.
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharm. 2007, 152, 1092–1101.
- Befort, K. Interactions of the opioid and cannabinoid systems in reward: Insights from knockout studies. Front. Pharm. 2015, 6.
- Meadows, A.; Lee, J.H.; Wu, C.-S.; Wei, Q.; Pradhan, G.; Yafi, M.; Lu, H.-C.; Sun, Y. Deletion of G-protein-coupled receptor 55 promotes obesity by reducing physical activity. Int. J. Obes. (London) 2016, 40, 417–424.
- Baskaran, P.; Krishnan, V.; Ren, J.; Thyagarajan, B. Capsaicin induces browning of white adipose tissue and counters obesity by activating TRPV1 channel-dependent mechanisms. Br. J. Pharm. 2016, 173, 2369–2389.
- Christie, S.; Wittert, G.A.; Li, H.; Page, A.J. Involvement of TRPV1 channels in energy homeostasis. Front. Endocrinol. 2018, 9, 420.
- Hryhorowicz, S.; Kaczmarek-Ryś, M.; Andrzejewska, A.; Staszak, K.; Hryhorowicz, M.; Korcz, A.; Słomski, R. Allosteric modulation of cannabinoid receptor 1—Current challenges and future opportunities. Int. J. Mol. Sci. 2019, 20, 5874.
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharm. 1995, 50, 83–90.
- Biernacki, M.; Skrzydlewska, E. Metabolizm endokannabinoidów. Postepy Hig. Med. Dosw. 2016, 70, 830–843.
- Porter, A.C.; Sauer, J.-M.; Knierman, M.D.; Becker, G.W.; Berna, M.J.; Bao, J.; Nomikos, G.G.; Carter, P.; Bymaster, F.P.; Leese, A.B.; et al. Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J. Pharm. Exp. 2002, 301, 1020–1024.
- Chen, G.; Pang, Z. Endocannabinoids and obesity. Vitam. Horm. 2013, 91, 325–368.
- Tsutsumi, T.; Matsuda, R.; Morito, K.; Kawabata, K.; Yokota, M.; Nikawadori, M.; Inoue-Fujiwara, M.; Kawashima, S.; Hidaka, M.; Yamamoto, T.; et al. Identification of human glycerophosphodiesterase 3 as an ecto phospholipase C that converts the G protein-coupled receptor 55 agonist lysophosphatidylinositol to bioactive monoacylglycerols in cultured mammalian cells. Biochim. Biophys Acta Mol. Cell Biol. Lipids 2020, 1865, 158761.
- Briand-Mésange, F.; Pons, V.; Allart, S.; Masquelier, J.; Chicanne, G.; Beton, N.; Payrastre, B.; Muccioli, G.G.; Ausseil, J.; Davignon, J.-L.; et al. Glycerophosphodiesterase 3 (GDE3) is a lysophosphatidylinositol-specific ectophospholipase C acting as an endocannabinoid signaling switch. J. Biol. Chem. 2020, 295, 15767–15781.
- Mackie, K. Distribution of Cannabinoid Receptors in the Central and Peripheral Nervous System; Handbook Experimental Pharmacology; Springer: Berlin, Germany, 2005; pp. 299–325.
- Perkins, J.M.; Davis, S.N. Endocannabinoid System overactivity and the metabolic syndrome: Prospects for treatment. Curr. Diab. Rep. 2008, 8, 12–19.
- Kendall, D.A.; Yudowski, G.A. Cannabinoid receptors in the central nervous system: Their signaling and roles in disease. Front. Cell Neurosci. 2017, 10.
- Matias, I.; Di Marzo, V. Endocannabinoids and the control of energy balance. Trends Endocrinol. Metab. 2007, 18, 27–37.
- Onaivi, E.S.; Ishiguro, H.; Gong, J.-P.; Patel, S.; Perchuk, A.; Meozzi, P.A.; Myers, L.; Mora, Z.; Tagliaferro, P.; Gardner, E.; et al. Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann. N. Y. Acad. Sci. 2006, 1074, 514–536.
- Zou, S.; Kumar, U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018, 19, 833.
- Ye, L.; Cao, Z.; Wang, W.; Zhou, N. New insights in cannabinoid receptor structure and signaling. Curr. Mol. Pharm. 2019, 12, 239–248.
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. An overview of the cannabinoid type 2 receptor system and its therapeutic potential. Curr. Opin. Anaesthesiol. 2018, 31, 407–414.
- Dhopeshwarkar, A.; Mackie, K. CB2 Cannabinoid receptors as a therapeutic target-what does the future hold? Mol. Pharm. 2014, 86, 430–437.
- Home-Gene-NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene (accessed on 2 August 2020).
- Kargl, J.; Balenga, N.; Parzmair, G.P.; Brown, A.J.; Heinemann, A.; Waldhoer, M. The cannabinoid receptor CB1 modulates the signaling properties of the lysophosphatidylinositol receptor GPR55. Biol. Chem. 2012, 287, 44234–44248.
- Atwood, B.K.; Mackie, K. CB2: A Cannabinoid receptor with an identity crisis. Br. J. Pharm. 2010, 160, 467–479.
- Kirkham, T.C.; Williams, C.M.; Fezza, F.; Di Marzo, V. Endocannabinoid levels in rat limbic forebrain and hypothalamus in relation to fasting, feeding and satiation: Stimulation of eating by 2-arachidonoyl glycerol. Br. J. Pharm. 2002, 136, 550–557.
- Matias, I.; Gonthier, M.-P.; Orlando, P.; Martiadis, V.; De Petrocellis, L.; Cervino, C.; Petrosino, S.; Hoareau, L.; Festy, F.; Pasquali, R.; et al. Regulation, function, and dysregulation of endocannabinoids in models of adipose and beta-pancreatic cells and in obesity and hyperglycemia. J. Clin. Endocrinol. Metab. 2006, 91, 3171–3180.
- Gary-Bobo, M.; Elachouri, G.; Scatton, B.; Le Fur, G.; Oury-Donat, F.; Bensaid, M. The cannabinoid CB1 receptor antagonist rimonabant (SR141716) inhibits cell proliferation and increases markers of adipocyte maturation in cultured mouse 3T3 F442A preadipocytes. Mol. Pharm. 2006, 69, 471–478.
- Pagano, C.; Pilon, C.; Calcagno, A.; Urbanet, R.; Rossato, M.; Milan, G.; Bianchi, K.; Rizzuto, R.; Bernante, P.; Federspil, G.; et al. The endogenous cannabinoid system stimulates glucose uptake in human fat cells via phosphatidylinositol 3-kinase and calcium-dependent mechanisms. J. Clin. Endocrinol. Metab. 2007, 92, 4810–4819.
- Silvestri, C.; Di Marzo, V. The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab. 2013, 17, 475–490.
- Cota, D.; Marsicano, G.; Tschöp, M.; Grübler, Y.; Flachskamm, C.; Schubert, M.; Auer, D.; Yassouridis, A.; Thöne-Reineke, C.; Ortmann, S.; et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J. Clin. Investig. 2003, 112, 423–431.
- Bajzer, M.; Olivieri, M.; Haas, M.K.; Pfluger, P.T.; Magrisso, I.J.; Foster, M.T.; Tschöp, M.H.; Krawczewski-Carhuatanta, K.A.; Cota, D.; Obici, S. Cannabinoid receptor 1 (CB1) antagonism enhances glucose utilisation and activates brown adipose tissue in diet-induced obese mice. Diabetologia 2011, 54, 3121–3131.
- Verty, A.N.A.; Allen, A.M.; Oldfield, B.J. The Effects of rimonabant on brown adipose tissue in rat: Implications for energy expenditure. Obesity 2009, 17, 254–261.
- Eriksson, O.; Mikkola, K.; Espes, D.; Tuominen, L.; Virtanen, K.; Forsback, S.; Haaparanta-Solin, M.; Hietala, J.; Solin, O.; Nuutila, P. The cannabinoid receptor-1 is an imaging biomarker of brown adipose tissue. J. Nucl. Med. 2015, 56, 1937–1941.
- Boon, M.R.; Kooijman, S.; van Dam, A.D.; Pelgrom, L.R.; Berbée, J.F.P.; Visseren, C.A.R.; Aggele, R.C.v.; van den Hoek, A.M.; Sips, H.C.M.; Lombès, M.; et al. Peripheral cannabinoid 1 receptor blockade activates brown adipose tissue and diminishes dyslipidemia and obesity. FASEB J. 2014, 28, 5361–5375.
- Lahesmaa, M.; Eriksson, O.; Gnad, T.; Oikonen, V.; Bucci, M.; Hirvonen, J.; Koskensalo, K.; Teuho, J.; Niemi, T.; Taittonen, M.; et al. Cannabinoid type 1 receptors are upregulated during acute activation of brown adipose tissue. Diabetes 2018, 67, 1226–1236.
- Woods, S.C. Role of the endocannabinoid system in regulating cardiovascular and metabolic risk factors. Am. J. Med. 2007, 120, S19–S25.
- Osei-Hyiaman, D.; DePetrillo, M.; Pacher, P.; Liu, J.; Radaeva, S.; Bátkai, S.; Harvey-White, J.; Mackie, K.; Offertáler, L.; Wang, L.; et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Investig. 2005, 115, 1298–1305.
- Medina, J.; Fernández-Salazar, L.I.; García-Buey, L.; Moreno-Otero, R. Approach to the pathogenesis and treatment of nonalcoholic steatohepatitis. Diabetes Care 2004, 27, 2057–2066.
- Jeong, W.; Osei-Hyiaman, D.; Park, O.; Liu, J.; Bátkai, S.; Mukhopadhyay, P.; Horiguchi, N.; Harvey-White, J.; Marsicano, G.; Lutz, B.; et al. Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab. 2008, 7, 227–235.
- Ruby, M.A.; Nomura, D.K.; Hudak, C.S.S.; Barber, A.; Casida, J.E.; Krauss, R.M. Acute overactive endocannabinoid signaling induces glucose intolerance, hepatic steatosis, and novel cannabinoid receptor 1 responsive genes. PLoS ONE 2011, 6, e26415.
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating amp-activated protein kinase. Nat. Med. 2002, 8, 1288–1295.
- Trebicka, J.; Racz, I.; Siegmund, S.V.; Cara, E.; Granzow, M.; Schierwagen, R.; Klein, S.; Wojtalla, A.; Hennenberg, M.; Huss, S.; et al. Role of cannabinoid receptors in alcoholic hepatic injury: Steatosis and fibrogenesis are increased in CB2 receptor-deficient mice and decreased in CB1 receptor knockouts. Liver Int. 2011, 31, 860–870.
- Liu, Y.L.; Connoley, I.P.; Wilson, C.A.; Stock, M.J. Effects of the cannabinoid CB1 receptor antagonist SR141716 on oxygen consumption and soleus muscle glucose uptake in Lep(Ob)/Lep(Ob) mice. Int. J. Obes. (London) 2005, 29, 183–187.
- Song, D.; Bandsma, R.H.J.; Xiao, C.; Xi, L.; Shao, W.; Jin, T.; Lewis, G.F. Acute cannabinoid receptor type 1 (CB1R) modulation influences insulin sensitivity by an effect outside the central nervous system in mice. Diabetologia 2011, 54, 1181–1189.
- Juan-Picó, P.; Fuentes, E.; Bermúdez-Silva, F.J.; Javier Díaz-Molina, F.; Ripoll, C.; Rodríguez de Fonseca, F.; Nadal, A. Cannabinoid receptors regulate Ca(2+) signals and insulin secretion in pancreatic beta-cell. Cell Calcium 2006, 39, 155–162.
- Starowicz, K.M.; Cristino, L.; Matias, I.; Capasso, R.; Racioppi, A.; Izzo, A.A.; Di Marzo, V. Endocannabinoid dysregulation in the pancreas and adipose tissue of mice fed with a high-fat diet. Obesity (Silver Spring) 2008, 16, 553–565.
- Gómez, R.; Navarro, M.; Ferrer, B.; Trigo, J.M.; Bilbao, A.; Arco, I.D.; Cippitelli, A.; Nava, F.; Piomelli, D.; de Fonseca, F.R. A peripheral mechanism for CB1 cannabinoid receptor-dependent modulation of feeding. J. Neurosci. 2002, 22, 9612–9617.
- Coutts, A.A.; Izzo, A.A. The gastrointestinal pharmacology of cannabinoids: An update. Curr. Opin. Pharm. 2004, 4, 572–579.
- Pang, Z.; Wu, N.N.; Zhao, W.; Chain, D.C.; Schaffer, E.; Zhang, X.; Yamdagni, P.; Palejwala, V.A.; Fan, C.; Favara, S.G.; et al. The central cannabinoid CB1 receptor is required for diet-induced obesity and rimonabant’s antiobesity effects in mice. Obesity (Silver Spring) 2011, 19, 1923–1934.
- DiPatrizio, N.V.; Astarita, G.; Schwartz, G.; Li, X.; Piomelli, D. Endocannabinoid signal in the gut controls dietary fat intake. Proc. Natl. Acad. Sci. USA 2011, 108, 12904–12908.
- Argueta, D.A.; DiPatrizio, N.V. Peripheral endocannabinoid signaling controls hyperphagia in western diet-induced obesity. Physiol. Behav. 2017, 171, 32–39.
- Di Marzo, V.; Matias, I. Endocannabinoid control of food intake and energy balance. Nat. Neurosci. 2005, 8, 585–589.
- Horvath, T.L. Endocannabinoids and the regulation of body fat: The smoke is clearing. J. Clin. Investig. 2003, 112, 323–326.
- Ohno-Shosaku, T.; Tanimura, A.; Hashimotodani, Y.; Kano, M. Endocannabinoids and Retrograde modulation of synaptic transmission. Neuroscientist 2012, 18, 119–132.
- Kim, J.; Li, Y.; Watkins, B.A. Endocannabinoid signaling and energy metabolism: A target for dietary intervention. Nutrition 2011, 27, 624–632.
- Cardinal, P.; Bellocchio, L.; Clark, S.; Cannich, A.; Klugmann, M.; Lutz, B.; Marsicano, G.; Cota, D. Hypothalamic CB1 Cannabinoid receptors regulate energy balance in mice. Endocrinology 2012, 153, 4136–4143.
- Di Marzo, V.; Goparaju, S.K.; Wang, L.; Liu, J.; Bátkai, S.; Járai, Z.; Fezza, F.; Miura, G.I.; Palmiter, R.D.; Sugiura, T.; et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 2001, 410, 822–825.
- Myers, M.G.; Heymsfield, S.B.; Haft, C.; Kahn, B.B.; Laughlin, M.; Leibel, R.L.; Tschöp, M.H.; Yanovski, J.A. Challenges and opportunities of defining clinical leptin resistance. Cell Metab. 2012, 15, 150–156.
- Tam, J.; Cinar, R.; Liu, J.; Godlewski, G.; Wesley, D.; Jourdan, T.; Szanda, G.; Mukhopadhyay, B.; Chedester, L.; Liow, J.-S.; et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012, 16, 167–179.
- Bisogno, T.; Mahadevan, A.; Coccurello, R.; Chang, J.-W.; Allarà, M.; Chen, Y.; Giacovazzo, G.; Lichtman, A.; Cravatt, B.; Moles, A.; et al. A novel fluorophosphonate inhibitor of the biosynthesis of the endocannabinoid 2-arachidonoylglycerol with potential anti-obesity effects. Br. J. Pharm. 2013, 169, 784–793.
- Quarta, C.; Bellocchio, L.; Mancini, G.; Mazza, R.; Cervino, C.; Braulke, L.J.; Fekete, C.; Latorre, R.; Nanni, C.; Bucci, M.; et al. CB(1) Signaling in forebrain and sympathetic neurons is a key determinant of endocannabinoid actions on energy balance. Cell Metab. 2010, 11, 273–285.
- Alén, F.; Ramírez-López, M.T.; Gómez de Heras, R.; Rodríguez de Fonseca, F.; Orio, L. Cannabinoid receptors and cholecystokinin in feeding inhibition. Vitam. Horm. 2013, 92, 165–196.
- Bermudez-Silva, F.J.; Cardinal, P.; Cota, D. The role of the endocannabinoid system in the neuroendocrine regulation of energy balance. J. Psychopharmacol. 2012, 26, 114–124.
- Melis, T.; Succu, S.; Sanna, F.; Boi, A.; Argiolas, A.; Melis, M.R. The cannabinoid antagonist SR 141716A (Rimonabant) reduces the increase of extra-cellular dopamine release in the rat nucleus accumbens induced by a novel high palatable food. Neurosci. Lett. 2007, 419, 231–235.
- Janero, D.R. Cannabinoid-1 receptor (CB1R) blockers as medicines: Beyond obesity and cardiometabolic disorders to substance abuse/drug addiction with CB1R neutral antagonists. Expert Opin. Emerg. Drugs 2012, 17, 17–29.
- DiPatrizio, N.V.; Piomelli, D. The thrifty lipids: Endocannabinoids and the neural control of energy conservation. Trends Neurosci. 2012, 35, 403–411.
- Naughton, S.S.; Mathai, M.L.; Hryciw, D.H.; McAinch, A.J. Fatty acid modulation of the endocannabinoid system and the effect on food intake and metabolism. Int. J. Endocrinol. 2013, 2013, 361895.
- Jager, G.; Witkamp, R.F. The endocannabinoid system and appetite: Relevance for food reward. Nutr. Res. Rev. 2014, 27, 172–185.
- Carta, G.; Melis, M.; Pintus, S.; Pintus, P.; Piras, C.A.; Muredda, L.; Demurtas, D.; Di Marzo, V.; Banni, S.; Barbarossa, I.T. Participants with normal weight or with obesity show different relationships of 6-n-propylthiouracil (PROP) taster status with BMI and plasma endocannabinoids. Sci. Rep. 2017, 7, 1361.
- Marco, E.M.; Romero-Zerbo, S.Y.; Viveros, M.-P.; Bermudez-Silva, F.J. The role of the endocannabinoid system in eating disorders: Pharmacological implications. Behav. Pharm. 2012, 23, 526–536.

