 +1 credit
+1 credit
Video Upload Options
The rapid growth of atmospheric CO2 concentration by continuous consumption of fossil fuels is one of the main causes of global warming. Turning CO2 into fuels and chemicals through biotransformation offers a win-win strategy to both decrease atmospheric CO2 and efficiently exploit carbon resources. The overall efficiency of CO2 biotransformation in vitro and CO2 assimilation in vivo is generally determined by the biochemical properties of carboxylases. Herein, we summarized carboxylases based on catalytic mechanism and CO2 biotransformation in vitro and CO2 assimilation in vivo based on newly mined or designed carboxylases.
1. Carboxylases for CO2 Biotransformation
CO2 is a poor electrophile and usually exists as bicarbonate in an aqueous solution. Therefore, the carboxylation reaction often requires energy (adenosine triphosphate (ATP), nicotinamide adenine dinucleotide phosphate (NADPH), or ferredoxin) or the assistance of coenzymes (metal ion, ThDP, and prenylated flavin mononucleotide (prFMN), etc.) [24]. We divide carboxylases into seven categories: (1) Only divalent metal-dependent carboxylases, (2) ATP-dependent carboxylases, (3) redox equivalents-dependent carboxylases, (4) substrate-activated carboxylases, (5) ThDP-dependent carboxylases, (6) multi-enzyme complex constructed carboxylase, (7) prFMN-dependent carboxylases. Representative carboxylases are shown in Table 1.
Table 1. Representative carboxylases for CO2 fixation.
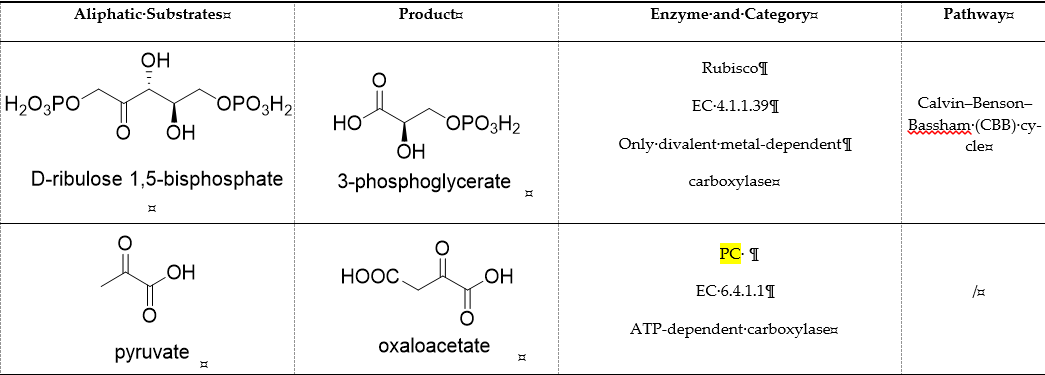
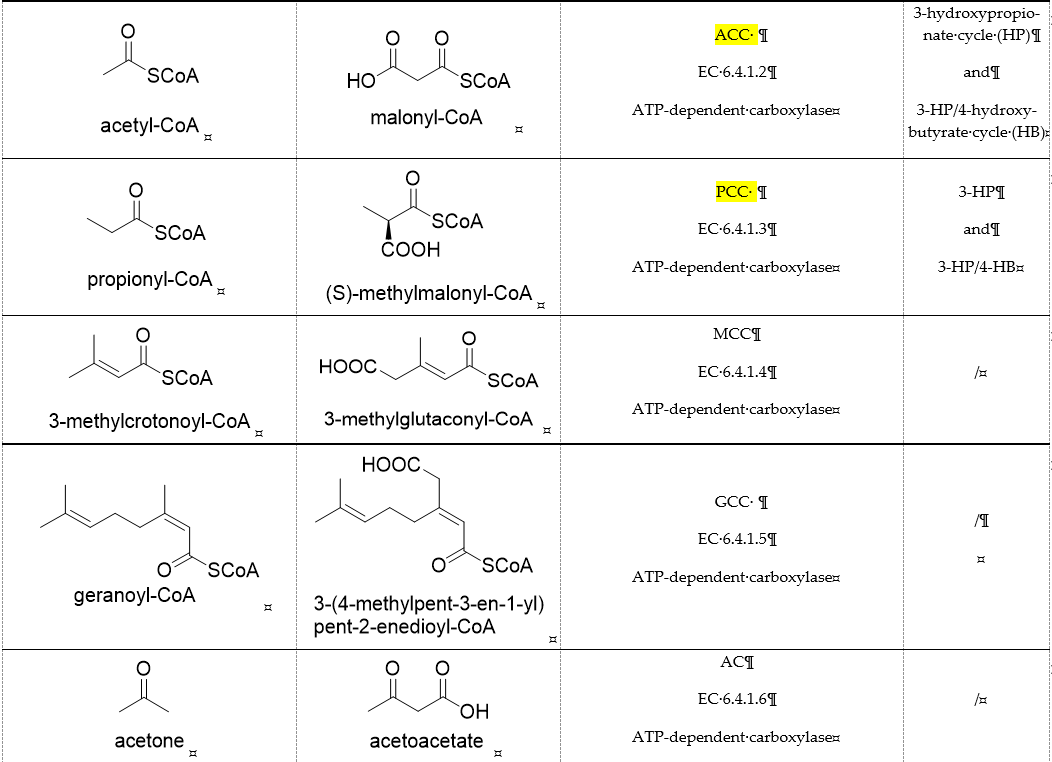
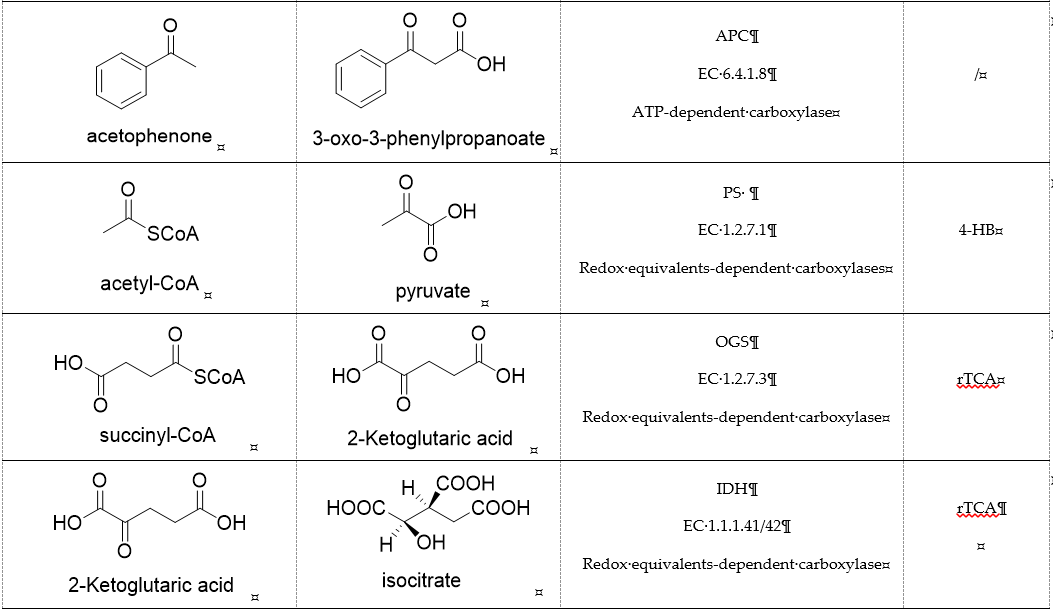
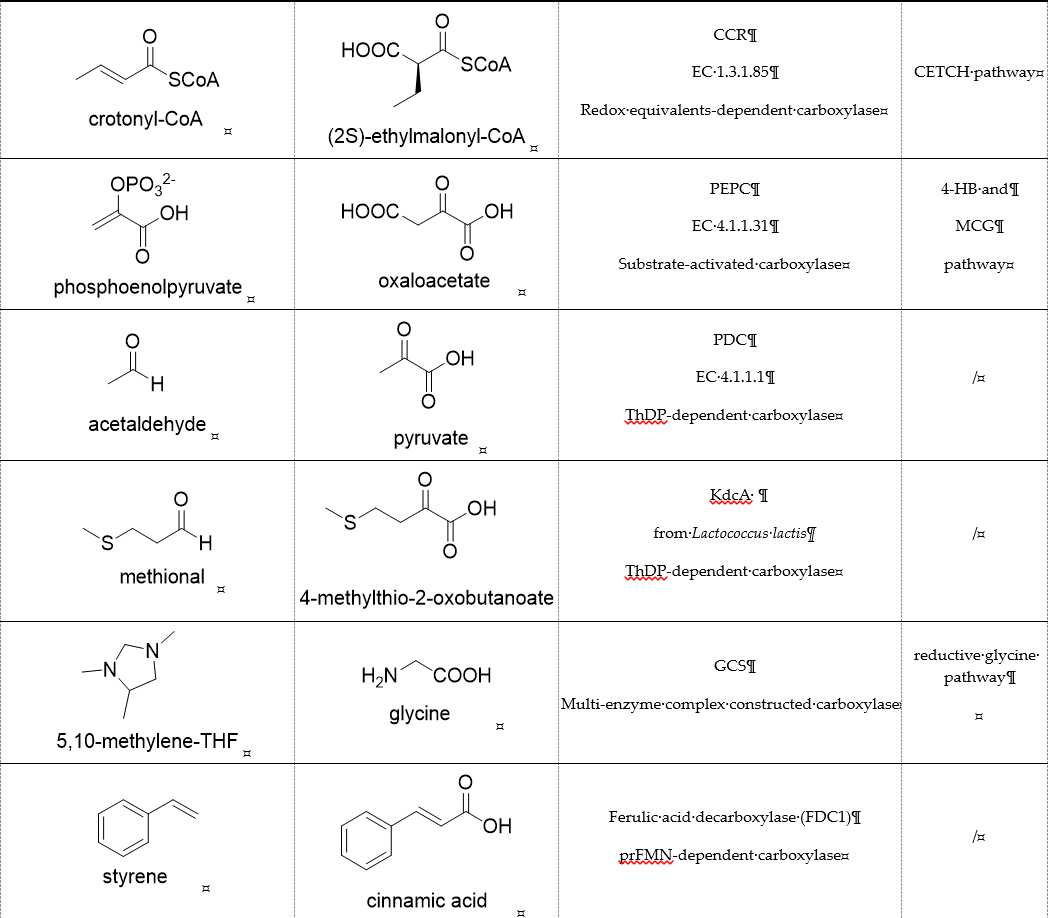
2. ATP-Dependent Carboxylases
Biotin-dependent carboxylases include pyruvate carboxylase (PC, EC 6.4.1.1), acetyl-CoA carboxylase (ACC, EC 6.4.1.2), propionyl-CoA carboxylase (PCC, EC 6.4.1.3), 3-methylcrotonoyl-CoA carboxylase (MCC, EC 6.4.1.4), and geranoyl-CoA carboxylase (GCC, EC 6.4.1.5). They are widely distributed in nature and can be found in archaea, bacteria, algae, fungi, plants, and animals[1]. The catalytic process of biotin-dependent carboxylases can be divided into two steps (Figure 2). First, the biotin carboxylase (BC) domain catalyzes the ATP-dependent carboxylation of the N1′ atom of the biotin cofactor, using bicarbonate as the CO2 donor. Second, the carboxyltransferase (CT) domain transfers the CO2 from carboxy-biotin to the substrates[2][3][4]. The site for carboxylation is on the α-carbon of saturated substrates (pyruvate, acetyl-CoA, and propionyl-CoA) or the γ-carbon of α, β-unsaturated substrates (3-methylcrotonyl-CoA, geranyl-CoA). Acetyl-CoA carboxylase and propionyl-CoA carboxylase are two carboxylases of 3-hydroxypropionate/malyl-CoA cycle and 3-hydroxypropionate/4-hydroxybutyrate cycle[5][6].
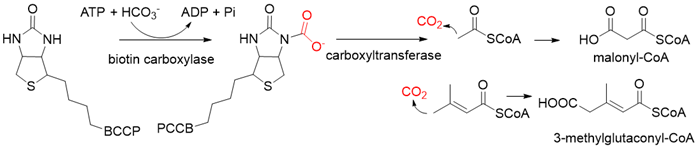
Figure 2. The catalytic process of biotin-dependent carboxylases[2][3][4].
Acetone carboxylases (AC, EC 6.4.1.6) are soluble cytoplasmic enzymes, and can be found in many species of aerobic, anaerobic phototrophic bacteria, and even microaerobic gastric human pathogenic species Helicobacter pylori. They catalyze the carboxylation of acetone to form acetoacetate at the expense of ATP[7]. There are two different types of acetone carboxylases. One requires 2 ATP equivalents as an energy supply for the carboxylation reaction, while another requires 4 ATP equivalents. The main difference in catalytic mechanism lies in the processes of substrate activation. The catalytic mechanism proposed for acetone carboxylase of Xanthobacter/Rhodobacter is that one ATP is sequentially hydrolyzed to ADP and AMP to activate acetone and bicarbonate, respectively. While the catalytic mechanism of acetone carboxylase from Aromatoleum is that 2 ATP are hydrolyzed to 2 AMP to active two substrates[8]. Acetoacetate can be activated by a CoA ligase to form acetoacetyl-CoA, which is cleaved to form 2 acetyl-CoA by thiolase. Therefore, a new pathway from isopropanol and CO2 to acetyl-CoA can be constructed. Acetophenone carboxylase (APC, EC 6.4.1.8) catalyzes the carboxylation of acetophenone to benzoylacetate[9]. Different from the above two activation processes of acetone carboxylases, acetophenone and bicarbonate are all activated by hydrolyzing ATP to ADP.
3. Redox Equivalents-Dependent Carboxylases
Pyruvate synthase (PS, EC 1.2.7.1) and 2-oxoglutarate synthase (OGS, EC 1.2.7.3) are a class of enzymes sharing a similar catalytic mechanism[10][11]. They belong to strictly anaerobic enzymes and show low catalytic activity. Acetyl-CoA can be reductively carboxylated by pyruvate synthase at the expense of two equivalents of ferredoxin to generate pyruvate. Similarly, succinyl-CoA can be converted to 2-oxoglutarate by 2-oxoglutarate synthase[12]. Different from the above two enzymes, isocitrate dehydrogenase (IDH, EC 1.1.1.41/42) converts 2-oxoglutarate to isocitrate at the expense of NAD(P)H[13]. The carboxylation process of isocitrate dehydrogenase is assumed to proceed via the enolate intermediate of 2-oxoglutarate, which is formed with the assistance of divalent metal ions Mg2+ or Mn2+. After the addition of CO2, the unstable keto-tricarboxylic acid intermediate is immediately reduced by NAD(P)H to yield stable isocitrate (Figure 3A). 2-oxoglutarate synthase and isocitrate dehydrogenase are carboxylases of the reductive tricarboxylic acid (rTCA) cycle.
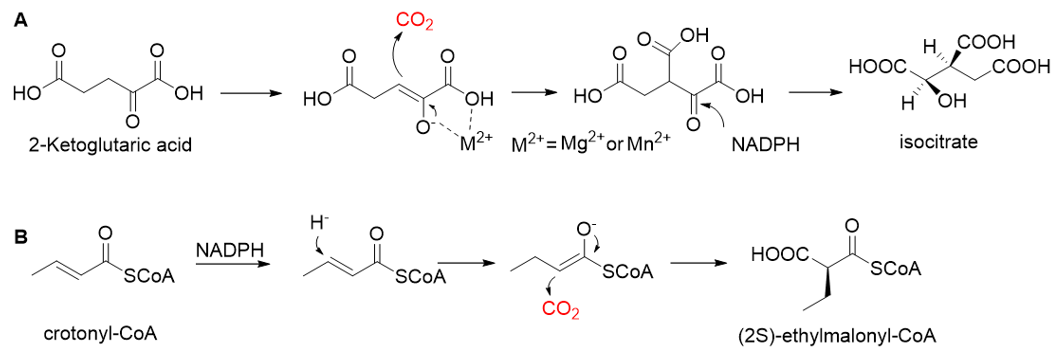
Figure 3. The catalytic mechanism of isocitrate dehydrogenase and crotonyl-CoA carboxylase/reductase. (A) The catalytic mechanism of isocitrate dehydrogenase[13]. (B) The catalytic mechanism of crotonyl-CoA carboxylase/reductase[14].
Enoyl-CoA carboxylases/reductases (ECRs) are a class of carboxylases that exist in secondary metabolism, as well as in central carbon metabolism of α-proteobacteria and Streptomycetes[14]. The best-studied ECR is crotonyl-CoA carboxylase/reductase (CCR, EC 1.3.1.85) that catalyzes NADPH-dependent reductive carboxylation of crotonyl-CoA into (2S)-ethylmalonyl-CoA. The mechanism of CCR is assumed to proceed via nucleophilic hydride attack at β-carbon of the enoyl-CoA ester; the forming enolate is trapped by CO2 to generate (2S)-ethylmalonyl-CoA (Figure 3B). Recently, combining experimental biochemistry, protein crystallography, and advanced computer simulations, Gabriele M. M. Stoffel et al. determined the CO2-binding residues at the active site of crotonyl-CoA carboxylase/reductase from Kitasatospora setae[15]. Propionyl-CoA synthase from Erythrobacter sp. NAP1, as well as an acrylyl-CoA reductase from Nitrosopumilus maritimus, have almost no carboxylation activity. Based on the determined CO2-binding residues, they used rational design to engineer two enzymes into carboxylases by increasing interactions of the proteins with CO2 and suppressing diffusion of water to the active site[16].
Relative to Rubisco, CCR is oxygen-insensitive, does not react with O2, requires only the NADPH, and catalyzes CO2 fixation with higher efficiency (Kcat/Km = 1642.6 s−1 mM−1)[17][18]. All of these characteristics make CCR a good candidate enzyme for the fixation of CO2. Based on CCR, Tobias J. Erb research group constructed a crotonyl-CoA/ethylmalonyl-CoA/hydroxybutyryl-CoA (CETCH) cycle in vitro[19], which consists of 17 enzymes and can convert CO2 into organic molecules at a rate of 5 nanomoles of CO2 per minute per milligram of protein (Figure 4). Recently, they successfully encapsulated thylakoids isolated from the spinach plant along with all enzymes of the CETCH pathway within water-in-oil droplets[20]. The encapsulated system could use light energy to produce glycolate from CO2, while also phosphorylating ADP to ATP.
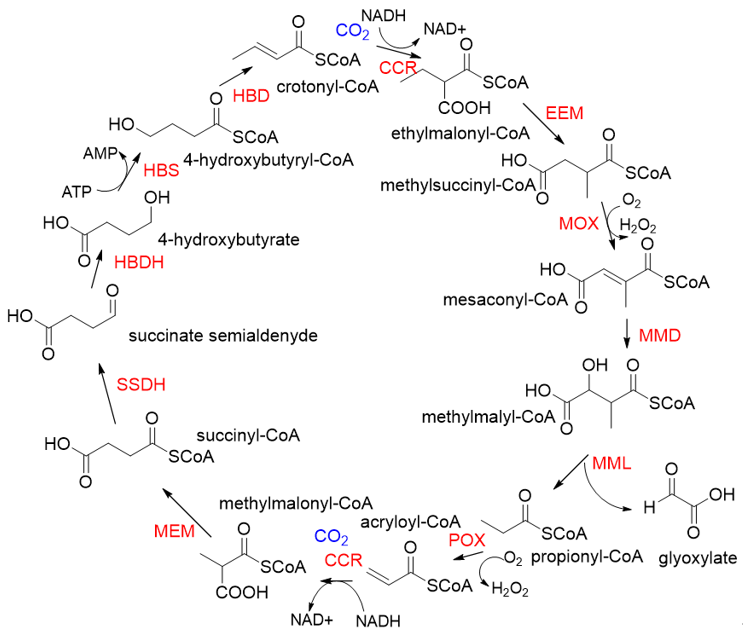
Figure 4. The crotonyl-CoA/ethylmalonyl-CoA/hydroxybutyryl-CoA cycle pathway (CETCH pathway)[19]. CCR, crotonyl-CoA carboxylase/reductase; EEM, ethylmalonyl-CoA epimerase, and mutase; MOX, methylsuccinyl-CoA oxidase; MMD, methylmalyl-CoA dehydratase; MML, methylmalyl-CoA lyase; POX, propionyl-CoA oxidase; MEM, methylmalonyl-CoA epimerase, and mutase; SSDH, succinate semialdehyde dehydrogenase; HBDH, 4-hydroxybutyrate dehydrogenase; HBS, 4-hydroxybutyryl-CoA synthetase; HBD, 4-hydroxybutyryl-CoA dehydratase.
4. Substrate-Activated Carboxylases
Phosphoenolpyruvate (PEP) carboxylase (PEPC, EC 4.1.1.31) catalyzes the irreversible carboxylation of PEP to form oxaloacetate (OAA) using Mg2+ or Mn2+ as a cofactor[21]. This kind of enzyme is present in most photosynthetic organisms[22]. PEPC is used to replenish intermediates of the TCA cycle for amino acid biosynthesis, or to shuttle CO2 between the mesophyll and bundle sheath cells in C4 plants. The catalytic mechanism of PEPC has been well studied (Figure 5). First, bicarbonate act as a nucleophile to attack phosphate groups in PEP, yielding carboxyphosphate and enolates of pyruvate, which is stabilized by metal ions Mn2+. Next, carboxyphosphate decomposes into inorganic phosphate and CO2, which is attacked by enolates of pyruvate to form OAA[23].
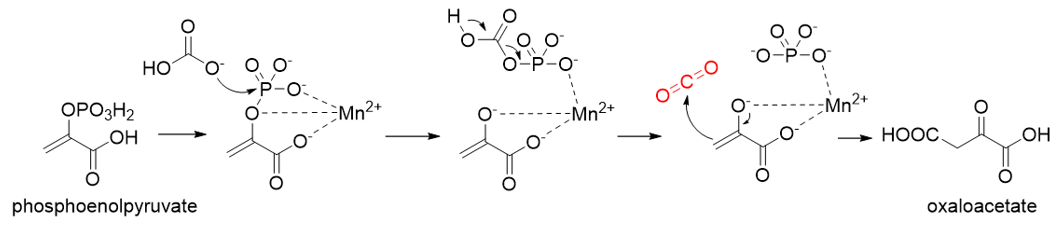
Figure 5. The catalytic mechanism of phosphoenolpyruvate carboxylase[23].
PEPC is known to be one of the most active carboxylases (Kcat/Km = 23,792 s−1 mM−1)[24]. Based on PEPC, Hong Yu et al. constructed a synthetic malyl-CoA-glycerate (MCG) pathway[25], which is capable of converting one C3 sugar to two acetyl-CoA via fixation of one CO2 equivalent, or assimilating glyoxylate, a photorespiration intermediate, to produce acetyl-CoA without carbon loss (Figure 6). Coupling the MCG pathway with the CBB cycle, photosynthetic organisms utilize only 5.5 ATP and 1.5 Rubisco turnovers to produce one acetyl-CoA from CO2 equivalents, while the native pathway requires 7 ATP and 3 Rubisco turnovers. When transferring the MCG pathway into a photosynthetic organism Synechococcus elongates PCC7942, the intracellular acetyl-CoA level increased, and bicarbonate assimilation was improved by roughly 2-fold.
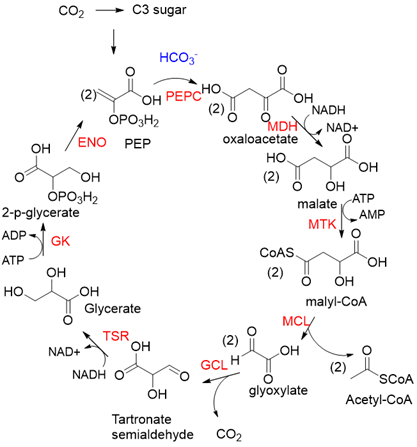
Figure 6. The synthetic malyl-CoA-glycerate (MCG) pathway[25]. PEPC, PEP carboxylase; MDH, malate dehydrogenase; MTK, malate thiokinase; MCL, malyl-CoA lyase; GCL, glyoxylate carboligase; TSR, tartronate semialdehyde reductase; GK, glycerate kinase; ENO, enolase.
5. ThDP-Dependent Carboxylases
Pyruvate decarboxylase (PDC, EC 4.1.1.1) is a key enzyme of carbon metabolism at the branching point between aerobic respiration and anaerobic alcoholic fermentation, and can be found in some bacteria, yeasts, and plants[26]. PDC catalyzes the decarboxylation of pyruvate by using ThDP and Mg2+ as cofactors (Figure 7). This enzyme has been successfully applied to yield pyruvic acid through the reverse carboxylation reaction. To favor the carboxylation, high pH and high bicarbonate concentration are needed[27]. In addition to using a high concentration of bicarbonate solution as a CO2 source, elevated CO2 pressure is also an effective way to drive the direction of carboxylation. Combining branched-chain α-keto acid decarboxylase (KdcA) from Lactococcus lactis with transaminase or amino acid dehydrogenase, Julia Martin et al. achieved the synthesis of L-methionine from the abundant industrial intermediate methional under a 2 bar CO2 atmosphere[28].
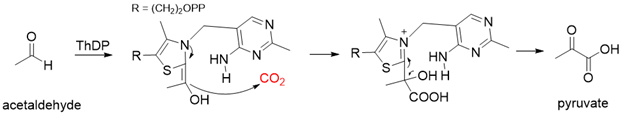
Figure 7. The catalytic mechanism of pyruvate decarboxylase[26][27].
6. Multi-Enzyme Complex Constructed Carboxylase
The glycine cleavage system (GCS) is common among many organisms because of its involvement in glycine and serine catabolism[29]. The GCS converts glycine to CO2, NH4+, and methylene-THF. GCS is composed of four proteins, a carrier protein, and three enzymes. They are lipoic acid-containing protein (GcvH), glycine dehydrogenase (GcvP), aminomethyltransferase (GcvT), and lipoamide dehydrogenase (Lpd), respectively. The glycine cleavage process can be divided into three steps. The first step is the decarboxylation of glycine by the glycine dehydrogenase. The decarboxylated moiety is then further degraded by the aminomethyl transferase with the aid of tetrahydrofolate. The last step is the reoxidation of the two sulfhydryl groups to form lipoic acid-generating NADH by dihydrolipomide dehydrogenase. Two sulfhydryl groups or lipoate attached to the lipoic acid-containing protein act as intermediate shuttles.
Now, GCS is confirmed to be reversible (rGCS) and can condense the C1 moiety of methylene-THF with CO2 and ammonia to produce glycine[30], which shows great application potential for reductive glycine pathway (RGP). Arren Bar-Even’s research group has made a series of encouraging progress[31][32]. Especially, they redesigned the central carbon metabolism of the model bacterium E. coli for growth on one-carbon compounds (formate and methanol) using the RGP[32] (Figure 8). Recently, Irene Sánchez-Andrea et al. demonstrated that sulfate-reducing bacterium Desulfovibrio desulfuricans (strain G11) could grow autotrophically via the RGP using hydrogen and sulfate as energy substrates[33]. This work first demonstrates that autotrophic microbial growth can be fully supported by RGP, which is a highly ATP-efficient CO2 fixation pathway.
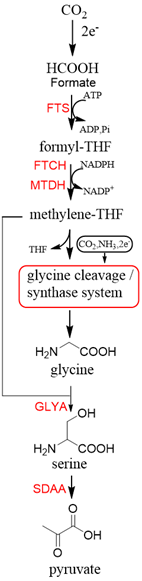
Figure 8. The reductive glycine pathway (RGP)[32]. FTS, formyl-THF synthase; FTCH, formyl-THF cyclohydrolase; MTDH, methylene-THF dehydrogenase; GLYA, L-serine hydroxymethyltransferase; SDAA, L-serine dehydratase.
7. prFMN-Dependent Carboxylases
prFMN-dependent decarboxylases catalyze the non-oxidative reversible decarboxylation of aromatic substrates, and play a pivotal role in bacterial ubiquinone (coenzyme Q) biosynthesis and microbial biodegradation of aromatic compounds[34][35]. The prFMN cofactor is provided by an associated prenyltransferase (UbiX), which extends the isoalloxazine FMN ring system through prenylation with a fourth non-aromatic ring. The catalytically active iminium species of the cofactor (prFMNiminium) is obtained by oxidizing the reduced prFMN with O2. There are two different catalytic reaction mechanisms for the prFMN-assisted (de)carboxylation reaction. For α, β-unsaturated carboxylic acids, the reaction proceeds through the intermolecular 1, 3-dipolar cycloaddition step. While for protocatechuic acid-type substrates, the electrophilic character of the iminium ion of prFMNiminium enables reversible (de)carboxylation via a mono-covalently bound quinoid–cofactor intermediate. prFMN-dependent decarboxylases encompass a wide range of substrates[36], including non-aromatic α, β-unsaturated (acrylic) acid derivatives, catechol, and 4-hydroxybenzoic acid derivatives, polycyclic aromatic hydrocarbons (PAHs), and heterocyclic substrates. Recently, combining ferulic acid decarboxylase (FDC, prFMN-dependent) with carboxylic acid reductase (CAR), alcohol dehydrogenase (ADH), or imine reductase (IRED), Godwin A. Aleku et al. designed cascade reactions to enable efficient functionalization of terminal alkenes to the corresponding aldehyde, alcohol, amide or amine derivatives through ambient CO2 fixation[37].
References
- Tong, L. Structure and function of biotin-dependent carboxylases. Cell Mol. Life Sci. 2013, 70, 863–891.
- Sarawut, J.; John, C.W. The Biotin Enzyme Family: Conserved Structural Motifs and Domain Rearrangements. Protein Pept. Sci. 2003, 4, 217–229.
- Maurice, M.; Reinhardt, L.; Surinya, K.H.; Attwood, P.V.; Wallace, J.C.; Cleland, W.W.; Rayment, I. Domain architecture of pyruvate carboxylase, a biotin-dependent multifunctional enzyme. Science 2007, 317, 1076–1079.
- Huang, C.S.; Sadre-Bazzaz, K.; Shen, Y.; Deng, B.; Zhou, Z.H.; Tong, L. Crystal structure of the α6β6 holoenzyme of propionyl-coenzyme A carboxylase. Nature 2010, 466, 1001–1005.
- Tong, L. Acetyl-coenzyme A carboxylase: Crucial metabolic enzyme and attractive target for drug discovery. Mol. life Sci. CMLS 2005, 62, 1784–1803.
- Wongkittichote, P.; Ah Mew, N.; Chapman, K.A. Propionyl-CoA carboxylase—A review. Genet. Metab. 2017, 122, 145–152.
- Heider, J.; Schuhle, K.; Frey, J.; Schink, B. Activation of Acetone and Other Simple Ketones in Anaerobic Bacteria. J. Mol. Microbiol. Biotechnol. 2016, 26, 152–164.
- Boyd, J.M.; Ensign, S.A. ATP-Dependent Enolization of Acetone by Acetone Carboxylase from Rhodobacter capsulatus. Biochemistry 2005, 44, 8543–8553.
- Weidenweber, S.; Schühle, K.; Demmer, U.; Warkentin, E.; Ermler, U.; Heider, J. Structure of the acetophenone carboxylase core complex: Prototype of a new class of ATP-dependent carboxylases/hydrolases. Rep. 2017, 7, 39674.
- Mai, X.; Adams, M.W. Characterization of a fourth type of 2-keto acid-oxidizing enzyme from a hyperthermophilic archaeon: 2-ketoglutarate ferredoxin oxidoreductase from Thermococcus litoralis. J. Bacteriol. 1996, 178, 5890–5896.
- Menon, A.L.; Hendrix, H.; Hutchins, A.; Verhagen, M.F.J.M.; Adams, M.W.W. The δ-Subunit of Pyruvate Ferredoxin Oxidoreductase from Pyrococcus furiosus Is a Redox-Active, Iron−Sulfur Protein: Evidence for an Ancestral Relationship with 8Fe-Type Ferredoxins. Biochemistry 1998, 37, 12838–12846.
- Ragsdale, S.W. Pyruvate Ferredoxin Oxidoreductase and Its Radical Intermediate. Rev. 2003, 103, 2333–2346.
- Soundar, S.; O’Hagan, M.; Fomulu, K.S.; Colman, R.F. Identification of Mn2+-binding Aspartates from α, β, and γ Subunits of Human NAD-dependent Isocitrate Dehydrogenase. Biol. Chem. 2006, 281, 21073–21081.
- Erb, T.J.; Berg, I.A.; Brecht, V.; Müller, M.; Fuchs, G.; Alber, B.E. Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: The ethylmalonyl-CoA pathway. Natl. Acad. Sci. USA 2007, 104, 10631.
- Stoffel, G.M.M.; Saez, D.A.; DeMirci, H.; Vögeli, B.; Rao, Y.; Zarzycki, J.; Yoshikuni, Y.; Wakatsuki, S.; Vöhringer-Martinez, E.; Erb, T.J. Four amino acids define the CO 2 binding pocket of enoyl-CoA carboxylases/reductases. Natl. Acad. Sci. USA 2019, 116, 13964.
- Bernhardsgrutter, I.; Schell, K.; Peter, D.M.; Borjian, F.; Saez, D.A.; Vohringer-Martinez, E.; Erb, T.J. Awakening the Sleeping Carboxylase Function of Enzymes: Engineering the Natural CO2-Binding Potential of Reductases. J. Am. Chem. Soc. 2019, 141, 9778–9782.
- Erb, T.J.; Brecht, V.; Fuchs, G.; Müller, M.; Alber, B.E. Carboxylation mechanism and stereochemistry of crotonyl-CoA carboxylase/reductase, a carboxylating enoyl-thioester reductase. Natl. Acad. Sci. USA 2009, 106, 8871.
- Peter, D.M.; Schada von Borzyskowski, L.; Kiefer, P.; Christen, P.; Vorholt, J.A.; Erb, T.J. Screening and Engineering the Synthetic Potential of Carboxylating Reductases from Central Metabolism and Polyketide Biosynthesis. Angewandte Chemie 2015, 54, 13457–13461.
- Schwander, T.; Schada von Borzyskowski, L.; Burgener, S.; Cortina, N.S.; Erb, T.J. A synthetic pathway for the fixation of carbon dioxide in vitro. Science 2016, 354, 900–904.
- Miller, T.; Beneyton, T.; Schwander, T.; Diehl, C.; Girault, M.; McLean, R.; Chotel, T.; Claus, P.; Cortina, N.; Baret, J.-C.; et al. Light-powered CO 2 fixation in a chloroplast mimic with natural and synthetic parts. Science 2020, 368, 649–654.
- Kai, Y.; Matsumura, H.; Izui, K. Phosphoenolpyruvate carboxylase: Three-dimensional structure and molecular mechanisms. Biochem. Biophys. 2003, 414, 170–179.
- Hatch, M.D. C4 photosynthesis: A unique elend of modified biochemistry, anatomy and ultrastructure. Biophys. Acta Rev. Bioenerg. 1987, 895, 81–106.
- Chollet, R.; Vidal, J.; O’Leary, M.H. PHOSPHOENOLPYRUVATE CARBOXYLASE: A Ubiquitous, Highly Regulated Enzyme in Plants. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 273–298.
- Cotton, C.A.; Edlich-Muth, C.; Bar-Even, A. Reinforcing carbon fixation: CO2 reduction replacing and supporting carboxylation. Opin. Biotechnol. 2018, 49, 49–56.
- Yu, H.; Li, X.; Duchoud, F.; Chuang, D.S.; Liao, J.C. Augmenting the Calvin-Benson-Bassham cycle by a synthetic malyl-CoA-glycerate carbon fixation pathway. Commun. 2018, 9, 2008.
- Kutter, S.; Wille, G.; Relle, S.; Weiss, M.S.; Hubner, G.; Konig, S. The crystal structure of pyruvate decarboxylase from Kluyveromyces lactis. Implications for the substrate activation mechanism of this enzyme. FEBS J. 2006, 273, 4199–4209.
- Miyazaki, M.; Shibue, M.; Ogino, K.; Nakamura, H.; Maeda, H. Enzymatic synthesis of pyruvic acid from acetaldehyde and carbon dioxide. Commun. 2001, 18, 1800–1801.
- Martin, J.; Eisoldt, L.; Skerra, A. Fixation of gaseous CO2 by reversing a decarboxylase for the biocatalytic synthesis of the essential amino acid l-methionine. Catal. 2018, 1, 555–561.
- Gonzales, J.N.; Matson, M.M.; Atsumi, S. Nonphotosynthetic Biological CO2 Biochemistry 2019, 58, 1470–1477.
- Tashiro, Y.; Hirano, S.; Matson, M.M.; Atsumi, S.; Kondo, A. Electrical-biological hybrid system for CO2 Metab. Eng. 2018, 47, 211–218.
- Yishai, O.; Bouzon, M.; Doring, V.; Bar-Even, A. In Vivo Assimilation of One-Carbon via a Synthetic Reductive Glycine Pathway in Escherichia coli. ACS Synth. Biol. 2018, 7, 2023–2028.
- Kim, S.; Lindner, S.N.; Aslan, S.; Yishai, O.; Wenk, S.; Schann, K.; Bar-Even, A. Growth of coli on formate and methanol via the reductive glycine pathway. Nat. Chem. Biol. 2020, 16, 538–545.
- Sánchez-Andrea, I.; Guedes, I.A.; Hornung, B.; Boeren, S.; Lawson, C.E.; Sousa, D.Z.; Bar-Even, A.; Claassens, N.J.; Stams, A.J.M. The reductive glycine pathway allows autotrophic growth of Desulfovibrio desulfuricans. Commun. 2020, 11, 5090.
- Payne, K.A.; White, M.D.; Fisher, K.; Khara, B.; Bailey, S.S.; Parker, D.; Rattray, N.J.; Trivedi, D.K.; Goodacre, R.; Beveridge, R.; et al. New cofactor supports α, β-unsaturated acid decarboxylation via 1,3-dipolar cycloaddition. Nature 2015, 522, 497–501.
- White, M.D.; Payne, K.A.; Fisher, K.; Marshall, S.A.; Parker, D.; Rattray, N.J.; Trivedi, D.K.; Goodacre, R.; Rigby, S.E.; Scrutton, N.S.; et al. UbiX is a flavin prenyltransferase required for bacterial ubiquinone biosynthesis. Nature 2015, 522, 502–506.
- Payer, S.E.; Faber, K.; Glueck, S.M. Non-Oxidative Enzymatic (De)Carboxylation of (Hetero)Aromatics and Acrylic Acid Derivatives. Synth. Catal. 2019, 361, 2402–2420.
- Aleku, G.A.; Saaret, A.; Bradshaw-Allen, R.T.; Derrington, S.R.; Titchiner, G.R.; Gostimskaya, I.; Gahloth, D.; Parker, D.A.; Hay, S.; Leys, D. Enzymatic C-H activation of aromatic compounds through CO2 fixation. Nat. Chem. Biol. 2020, 16, 1255–1260.

