 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | George W. Booz | + 2507 word(s) | 2507 | 2021-02-24 06:42:57 | | | |
| 2 | Rita Xu | -1078 word(s) | 1429 | 2021-03-03 07:27:12 | | |
Video Upload Options
Ischemic stroke is one of the most disabling diseases and a leading cause of death globally. Despite advances in medical care, the global burden of stroke continues to grow, as no effective treatments to limit or reverse ischemic injury to the brain are available. However, recent preclinical findings have revealed the potential role of transient receptor potential cation 6 (TRPC6) channels as endogenous protectors of neuronal tissue. Activating TRPC6 in various cerebral ischemia models has been found to prevent neuronal death, whereas blocking TRPC6 enhances sensitivity to ischemia. Evidence has shown that Ca2+ influx through TRPC6 activates the cAMP (adenosine 3’,5’-cyclic monophosphate) response element-binding protein (CREB), an important transcription factor linked to neuronal survival.
1. Introduction
In the USA, someone experiences a stroke every 40 s (https://www.cdc.gov/stroke/facts.htm; accessed January 2, 2021) or dies of a stroke every 4 min [1]. More than 795,000 people have a stroke every year, with 77% of these being first-time strokes [1]. In 2018, one of every six deaths in America from cardiovascular disease resulted from a stroke, making it the fifth leading cause of death in the USA. Some 87% of all strokes are ischemic strokes, in which blood flow to the brain is blocked, with the remainder classified as hemorrhagic due to rupture of a weakened blood vessel [1]. Moreover, stroke-related costs involving medicines, health care, and lost employment are staggering, totaling almost USD 50 billion between 2014 and 2015 [1].
Stroke is a leading cause of severe long-term disability, reducing mobility in more than half of the survivors 65 years old and over. Stroke is also the second leading cause of death globally [2]. Although the risk of stroke increases with age, it can occur at any age and in 2009, 34% of those hospitalized for stroke were younger than the age of 65 [3]. Despite advances in medical care, the global burden of stroke continues to grow [4]. Current thrombolytic therapy works for treating ischemic stroke, but only in a limited timeframe. No drugs are approved that enhance recovery, and thus, there is a great need to identify viable pharmacological targets. Furthermore, the current COVID-19 pandemic is directly increasing the incidence of ischemic injury and is worsening the incidence and prevalence of stroke in high-risk populations, as a result—in part—of increases in sedentary lifestyles associated with social distancing [5][6].
Stroke has two major types: hemorrhagic and ischemic, with the latter caused by cerebral embolism or thrombosis [7][8]. Despite varied etiologies for ischemia and hemorrhage, oxygen deprivation to neuronal tissue is a common mechanism. With hemorrhagic stroke, which is generally more severe [9], additional damage results from irritation and swelling from pressure build-up in surrounding tissues due to bleeding and blood breakdown products. In ischemic stroke, there is sudden occlusion of cerebral blood vessels, which leads to the interruption of, or reductions in blood supply to the brain tissue, resulting in extensive neuronal death [10]. This process involves a complex cascade of events at both the macro and microscopic levels, involving impaired vascular autoregulation, disruption of the blood–brain barrier (BBB), calcium overload-associated apoptosis, and neuronal death [11][12][13]. A key step is energy depletion from reduced blood flow, leading to Na+/K+ ATPase (sodium pump) failure, which causes cell membrane depolarization and glutamate release. Na+/K+ ATPase failure results in activation of proteases, kinases, and lipases, which contribute to tissue damage and necrosis. There is also a surge in phospholipase A2 activity that results in arachidonic acid (AA) release and enhanced free radical formation and lipid peroxidation [14][15]. Combined with non-neuronal (i.e., glial) cell activation, the neurovascular unit, consisting of astrocytes, endothelial cells and their BBB forming tight junctions, and pericytes, is impaired [16]. BBB disruption is further enhanced by neuronal glutamine release that activates endothelial N-methyl-D-aspartate (NMDA) receptor-mediated intracellular Ca2+ influx [17][18]. Glutamine is a major contributor to Ca2+ influx in neurons after ischemic stroke, and thus, also to the associated neurotoxicity.
Ischemic events may additionally activate Ca2+ influx via a non-glutamate pathway, including via transient receptor potential cation/canonical (TRPC) channels [19][20]. Increased cytosolic Ca2+ with ischemia can induce apoptosis and neuronal death by several means, including activation of calpains [11][19]. TRPC channels have been linked to vasospasm in hemorrhagic stroke [21]. Paradoxically one family member, TRPC6, has been linked to neuroprotection with ischemic stroke. It is reported that activating TRPC6 in a rat model of cerebral ischemia was shown to prevent neuronal death, whereas blocking TRPC6 enhanced sensitivity to ischemia [22]. One mechanism is that in patients treated for acute ischemic stroke, elevated expression levels in the peripheral blood of miR-488 and miR-135b, which were shown to target the TRPC6 gene, were identified as risk factors or associated with disease severity [23][24].
2. TRPC6
TRPC is a subfamily of TRP channels that are expressed in many cell types, including neurons [25][26]. These nonselective, cell membrane cation channels consist of seven members, TRPC 1–7, which depolarize cells via Na+ influx and also allow an influx of extracellular Ca2+, so as to regulate downstream cellular responses. Thus, this facilitates metabolism, membrane depolarization, gene expression, cell proliferation, and apoptosis [27]. Although all members are expressed in the brain and can promote nonselective Ca2+ entry, the spatial and temporal expression patterns of each are unclear. Based on structure–function relationships, the TRPC family is grouped into four subsets: TRPC1, TRPC2, TRPC3/6/7 and TRPC4/5. Besides functioning as homotetramers, combinations of different TRPC subunits can form heterotetrameric complexes, which may regulate responses to neuropeptides and neurotransmitters with different properties than homotetrameric TRPC channels [28]. For instance, TRPC6 has high sequence homology with TRPC3 and TRPC7 subunits, and may form a heterotetramer with TRPC3 [29].
TRPC6 possesses three conserved domains, namely, a pore-loop motif, four NH2 terminal ankyrin repeat domains (ARD), and a COOH-terminal TRP box motif (Figure 1) [30]. The ARD domains are thought to participate in channel heterodimerization and trafficking, whereas the TRP domain may be important for regulating binding with the cytoskeleton and translocation to the cell surface [30]. The pore-loop region is associated with an extracellular selectivity filter and an intracellular gate. Different TRPCs exhibit different Ca2+ and Na+ permeability (P) ratios [31]. The PCa/PNa ratio of TRPC6 is 5, compared to 1.6 for TRPC3 [32][33].
TRPC6 activation mediates changes in cytosolic Ca2+, which govern diverse critical cellular functions (Figure 1), such as contraction, apoptosis, neuroprotection, angiogenesis, and cytokine production. TRPC6 and other TRPCs can be activated by phospholipase C (PLC) by numerous stimulations, such as inflammation and ischemia-reperfusion (IR) injury [34]. Through activation of G-protein coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs), PLC can modulate TRPC channel activity by hydrolysis of phosphatidylinositol bisphosphate (PIP2) to diacylglycerol (DAG) and inositol trisphosphate (IP3) [35]. DAG activates TRPC3/6/7. IP3 causes Ca2+ release from internal stores, a process that triggers store-operated channel activation and may involve TRPC channels, although exactly how TRPC and store-operated channels interact is unclear [36][37]. Acting in a negative feedback manner, Ca2+ may reduce TRPC channel activity in synergy with protein kinase C (PKC), or via activation of calmodulin [38][39]. Additionally, TRPC3 and possibly TRPC6 may be activated as well in a β-arrestin-1-dependent manner [39][40][41]. These features make TRPC channels potential cellular sensors to respond to environmental changes by regulating intracellular Ca2+.
Structurally, TRPC6 is tethered directly to the cytoskeleton or extracellular matrix [42]. Studies on mouse embryonic fibroblasts and ventricular myocytes have suggested that TRPC6 may mediate the influx of Ca2+ in response to mechanical stress on the cell membrane [43][44]; however, TRPC6, expressed by various mammalian cell lines or in lipid bilayers, does not function as a mechanoreceptor [45][46]. Nonetheless, it may have evolved to discriminate different mechanical stimuli based on its interactions with the cytoskeleton or extracellular components. Therefore, TRPC6 may act as a secondary mechanoreceptor that contributes to the regulation of intracellular Ca2+ or depolarization of the membrane potential through inward Na+ and/or Ca2+ currents.
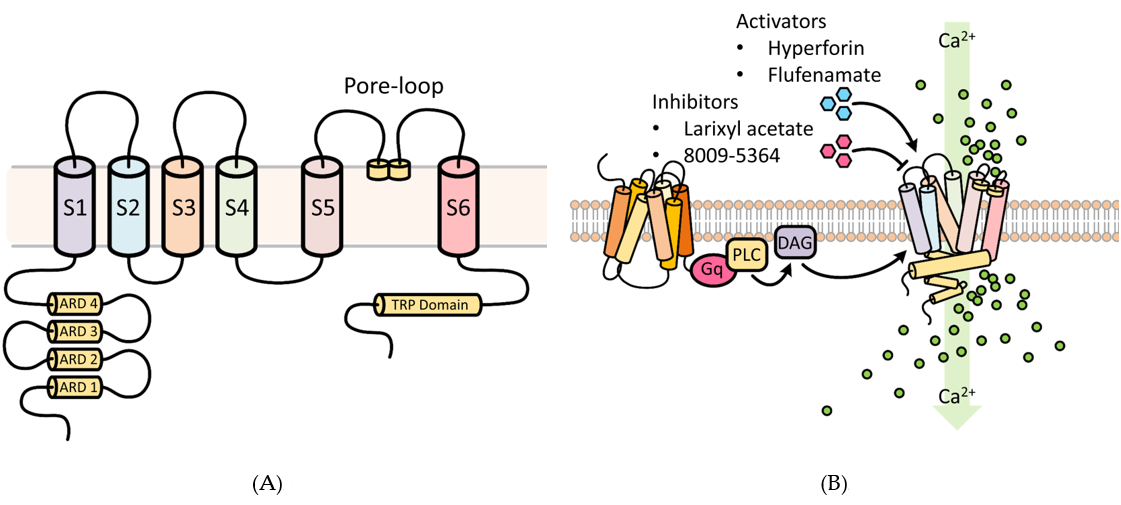
Figure 1. Functional aspects of transient receptor potential cation 6 (TRPC6) involved in Ca2+ cellular influx. (A) Structural features of the TRPC6 channel. TRPC6 possesses 6 membrane-spanning domains and 3 conserved domains, namely, a pore-loop motif, four NH2 terminal ankyrin repeat domains (ARD), and a COOH-terminal TRP box motif. The ARD domains participate in channel heterodimerization and trafficking, whereas the TRP domain regulates binding with the cytoskeleton and translocation to the cell surface. The pore-loop region (between the S5 and S6) is associated with an extracellular selectivity filter and an intracellular gate and allows for the passage of cations. ARD: ankyrin repeat domain; S1–S6 are the first to sixth transmembrane domains; TRP, transient receptor potential. (B) Regulators of TRPC6 channel activity. Receptor tyrosine kinases (RTKs) or G protein-coupled receptors (shown) increase TRPC6 activity by stimulating phospholipase C (PLC) to generate diacylglycerol (DAG). Several key experimental activators and inhibitors of TRPC6 are described in Section 6.
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596.
- Lopez, A.D.; Mathers, C.D.; Ezzati, M.; Jamison, D.T.; Murray, C.J. Global and regional burden of disease and risk factors, 2001: Systematic analysis of population health data. Lancet 2006, 367, 1747–1757.
- Hall, M.J.; Levant, S.; DeFrances, C.J. Hospitalization for stroke in U.S. hospitals, 1989-2009. NCHS Data Brief 2012, 5 , 1–8.
- Gorelick, P.B. The global burden of stroke: Persistent and disabling. Lancet Neurol. 2019, 18, 417–418.
- Fifi, J.T.; Mocco, J. COVID-19 related stroke in young individuals. Lancet Neurol. 2020, 19, 713–715.
- Jakobsson, J.; Malm, C.; Furberg, M.; Ekelund, U.; Svensson, M. Physical Activity During the Coronavirus (COVID-19) Pandemic: Prevention of a Decline in Metabolic and Immunological Functions. Sports Active Living 2020, 2, 57
- Grysiewicz, R.A.; Thomas, K.; Pandey, D.K. Epidemiology of ischemic and hemorrhagic stroke: Incidence, prevalence, mortality, and risk factors. Clin. 2008, 26, 871–895.
- Kim, S.M.; Jung, J.M.; Kim, B.J.; Lee, J.S.; Kwon, S.U. Cilostazol Mono and Combination Treatments in Ischemic Stroke: An Updated Systematic Review and Meta-Analysis. Stroke 2019, 50, 3503–3511.
- Andersen, K.K.; Olsen, T.S.; Dehlendorff, C.; Kammersgaard, L.P. Hemorrhagic and ischemic strokes compared: Stroke severity, mortality, and risk factors. Stroke 2009, 40, 2068–2072.
- Sekerdag, E.; Solaroglu, I.; Gursoy-Ozdemir, Y. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Neuropharmacol. 2018, 16, 1396–1415.
- Bano, D.; Nicotera, P. Ca2+ signals and neuronal death in brain ischemia. Stroke 2007, 38(2 Suppl), 674–676.
- Smith, W.S., Pathophysiology of focal cerebral ischemia: A therapeutic perspective. Vasc. Interv. Radiol. 2004, 15, S3–12.
- Shekhar, S.; Liu, R.; Travis, O.K.; Roman, R.J.; Fan, F. Cerebral Autoregulation in Hypertension and Ischemic Stroke: A Mini Review. Pharm. Sci. Exp. Pharmacol. 2017, 2017, 21–27.
- Durukan, A.; Tatlisumak, T. Acute ischemic stroke: Overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Biochem. Behav. 2007, 87, 179–197.
- Muralikrishna Adibhatla, R.; Hatcher, J.F. Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic. Biol. Med. 2006, 40, 376–387.
- Yang, C.; Hawkins, K.E.; Dore, S.; Candelario-Jalil, E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. J. Physiol. Cell Physiol. 2019, 316, C135–C153.
- Unterberg, A.W.; Stover, J.; Kress, B.; Kiening, K.L. Edema and brain trauma. Neuroscience 2004, 129, 1021–1029.
- Iadecola, C., Regulation of the cerebral microcirculation during neural activity: Is nitric oxide the missing link? Trends Neurosci. 1993, 16, 206–214.
- Huang, Q.; Wang, X.; Lin, X.; Zhang, J.; You, X.; Shao, A. The Role of Transient Receptor Potential Channels in Blood-Brain Barrier Dysfunction after Ischemic Stroke. Pharmacother. 2020, 131, 110647.
- Li, H.; Huang, J.; Du, W.; Jia, C.; Yao, H.; Wang, Y. TRPC6 inhibited NMDA receptor activities and protected neurons from ischemic excitotoxicity. Neurochem. 2012, 123, 1010–1018.
- Huang, J., TRPC Channels and Stroke. Exp. Med. Biol. 2017, 976, 61–71.
- Du, W.; Huang, J.; Yao, H.; Zhou, K.; Duan, B.; Wang, Y. Inhibition of TRPC6 degradation suppresses ischemic brain damage in rats. Clin. Invest. 2010, 120, 3480–3492.
- Liang, J.; Zhang, Z. Higher Peripheral Blood MiR-488 Level Predicts Poor Prognosis of Acute Ischemic Stroke. Lab. 2020, 66, PMID: 32658439.
- Yang, S.; Zhan, X.; He, M.; Wang, J.; Qiu, X. miR-135b levels in the peripheral blood serve as a marker associated with acute ischemic stroke. Ther. Med. 2020, 19, 3551–3558.
- Montell, C.; Birnbaumer, L.; Flockerzi, V.; Bindels, R.J.; Bruford, E.A.; Caterina, M.J.; Clapham, D.E.; Harteneck, C.; Heller, S.; Julius, D.; et al. A unified nomenclature for the superfamily of TRP cation channels. Cell 2002, 9, 229–231.
- Dietrich, A.; Chubanov, V.; Kalwa, H.; Rost, B.R.; Gudermann, T. Cation channels of the transient receptor potential superfamily: Their role in physiological and pathophysiological processes of smooth muscle cells. Ther. 2006, 112, 744–760.
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harbor Perspect. Biol. 2010, 2, a003962.
- Gualdani, R.; Gailly, P. How TRPC Channels Modulate Hippocampal Function. J. Mol. Sci. 2020, 21, PMID: 32658439
- Hirschler-Laszkiewicz, I.; Tong, Q.; Conrad, K.; Zhang, W.; Flint, W.W.; Barber, A.J.; Barber, D.L.; Cheung, J.Y.; Miller, B.A. TRPC3 activation by erythropoietin is modulated by TRPC6. Biol. Chem. 2009, 284, 4567–4581.
- Venkatachalam, K.; Montell, C. TRP channels. Rev. Biochem. 2007, 76, 387–417.
- Kamouchi, M.; Philipp, S.; Flockerzi, V.; Wissenbach, U.; Mamin, A.; Raeymaekers, L.; Eggermont, J.; Droogmans, G.; Nilius, B. Properties of heterologously expressed hTRP3 channels in bovine pulmonary artery endothelial cells. Physiol. 1999, 518, 345–358.
- Zitt, C.; Obukhov, A.G.; Strubing, C.; Zobel, A.; Kalkbrenner, F.; Luckhoff, A.; Schultz, G. Expression of TRPC3 in Chinese hamster ovary cells results in calcium-activated cation currents not related to store depletion. Cell Biol. 1997, 138, 1333–1341.
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263.
- Kelly, M.J.; Qiu, J.; Ronnekleiv, O.K. TRPCing around the hypothalamus. Neuroendocrinol. 2018, 51, 116–124.
- Moran, M.M.; Xu, H.; Clapham, D.E. TRP ion channels in the nervous system. Opin. Neurobiol. 2004, 14, 362–369.
- Nilius, B.; Owsianik, G.; Voets, T.; Peters, J.A. Transient receptor potential cation channels in disease. Rev. 2007, 87, 165–217.
- Thakore, P.; Earley, S. Transient Receptor Potential Channels and Endothelial Cell Calcium Signaling. Physiol. 2019, 9, 1249–1277.
- Putney, J.W.; Tomita, T. Phospholipase C signaling and calcium influx. Biol. Regul. 2012, 52, 152–164.
- Chen, X.; Sooch, G.; Demaree, I.S.; White, F.A.; Obukhov, A.G., Transient Receptor Potential Canonical (TRPC) Channels: Then and Now. Cells 2020, 9, 1983.
- Chai, Z.; Chen, Y.; Wang, C. beta-arrestin-1: Bridging GPCRs to active TRP channels. Channels (Austin) 2017, 11, 357–359.
- Liu, C.H.; Gong, Z.; Liang, Z.L.; Liu, Z.X.; Yang, F.; Sun, Y.J.; Ma, M.L.; Wang, Y.J.; Ji, C.R.; Wang, Y.H.; et al. Arrestin-biased AT1R agonism induces acute catecholamine secretion through TRPC3 coupling. Commun. 2017, 8, 14335.
- Patel, A.; Sharif-Naeini, R.; Folgering, J.R.; Bichet, D.; Duprat, F.; Honore, E. Canonical TRP channels and mechanotransduction: From physiology to disease states. Pflugers Archiv : Eur. J. Physiol. 2010, 460, 571–581.
- Dyachenko, V.; Husse, B.; Rueckschloss, U.; Isenberg, G. Mechanical deformation of ventricular myocytes modulates both TRPC6 and Kir2.3 channels. Cell Calcium 2009, 45, 38–54.
- Lei, L.; Lu, S.; Wang, Y.; Kim, T.; Mehta, D.; Wang, Y. The role of mechanical tension on lipid raft dependent PDGF-induced TRPC6 activation. Biomaterials 2014, 35, 2868–2877.
- Nikolaev, Y.A.; Cox, C.D.; Ridone, P.; Rohde, P.R.; Cordero-Morales, J.F.; Vasquez, V.; Laver, D.R.; Martinac, B. Mammalian TRP ion channels are insensitive to membrane stretch. Cell Sci. 2019, 132, PMID: 31722978
- Mederos y Schnitzler, M.; Storch, U.; Meibers, S.; Nurwakagari, P.; Breit, A.; Essin, K.; Gollasch, M.; Gudermann, T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 2008, 27, 3092–3103.

