 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Thomas Liehr | + 2262 word(s) | 2262 | 2021-03-02 08:01:26 | | | |
| 2 | Vivi Li | Meta information modification | 2262 | 2021-03-03 07:30:46 | | |
Video Upload Options
Repetitive DNA in humans is still widely considered to be meaningless, and variations within this part of the genome are generally considered to be harmless to the carrier. In contrast, for euchromatic variation, one becomes more careful in classifying inter-individual differences as meaningless and rather tends to see them as possible influencers of the so-called ‘genetic background’, being able to at least potentially influence disease susceptibilities. Here, the known ‘bad boys’ among repetitive DNAs are reviewed. Variable numbers of tandem repeats (VNTRs = micro- and minisatellites), small-scale repetitive elements (SSREs) and even chromosomal heteromorphisms (CHs) may therefore have direct or indirect influences on human diseases and susceptibilities. Summarizing this specific aspect here for the first time should contribute to stimulating more research on human repetitive DNA. It should also become clear that these kinds of studies must be done at all available levels of resolution, i.e., from the base pair to chromosomal level and, importantly, the epigenetic level, as well.
1. Introduction
In humans, like in other higher species, the genome of one individual never looks 100% alike to another one [1], even among those of the same gender or between monozygotic twins [2]. When comparing individuals, it seems to be a rule than an exception that there are many genetic differences that do not have obvious, meaning simply traceable, effects on the phenotype. Such genetic differences can be found at all levels of resolution when studying a genome, from the base pair to the chromosomal (i.e., cytogenetic) level and any other level in between [1]. Certainly, a species, including humans, is defined by the numbers of genes and chromosomes. However, while the normal chromosome number in humans has been determined to be 46,XX or 46,XY [3], the number and definition of ‘a gene’ are unclear, even in humans [4]. As nicely summarized in [4], studies on the human genome size in the 1990s suggested that the human genome contains 50,000–100,000 protein-coding genes (PTGs); the first sequence of the genome in 2001 contained ~25,000–30,000 PTGs. In 2004, 22,287 protein-coding genes and 34,214 transcripts were reported in the Ensembl human gene catalog. Since 2008, RNA-seq has further identified a sheer endless series of non-coding transcribed sequences, which are grouped into long non-coding RNAs (lncRNAs), antisense RNA and miscellaneous RNA. In 2018, ~20,000 protein-coding genes, ~15,000 pseudogenes and ~17,000–25,500 non-coding RNAs were identified [4]. In Table 1, the corresponding numbers are given as of 2021 [5][6][7][8]. Furthermore, there are variations in the euchromatic coding sequences of healthy individuals (i.e., different alleles), and much more variability has been described in non-coding sequences [1][2][9]. From an evolutionary standpoint, these differences are reserved for adaptations of a population to new environmental conditions [1][10]. However, the majority of repetitive DNA sequences have not been sequenced and/or are not identifiable by currently applied methods. According to a recent paper from 2020, there are still 783 unclosed sequence gaps dispersed over 150 Mb (GRCh38 assembly) [11]. Many of them are due to the technical limitations of present sequencing approaches, especially due to the limitations of data processing pipelines [11][12].
Table 1. Number of genes in the human genome as of 2021.
| Type | GENCODE [5] | NCBI [6] | Ensemble [7] | Genecards [8] |
|---|---|---|---|---|
| Total number of genes | 60,660 | 54,644 | 59,662 | 270,168 |
| Protein-coding genes | 19,962 | 20,203 | 20,448 | 20,916 |
| Genes that have more than one distinct translation | 13,685 * | 20,110 * | n.a. | n.a. |
| Non-coding genes | n.a. | 17,871 | 23,997 | 219,587 |
| lncRNA genes | 17,958 | n.a. | n.a. | 75,839 * |
| Small ncRNA/pir ncRNA genes | 7569 | n.a. | n.a. | 109,820 * |
| Pseudogenes | 14,761 | 15,067 | 15,217 | 21,888 |
| Others | 350 | 1503 | n.a. | 7777 |
The fields with * indicate that they have not been summarized into the total number of genes.
In humans, polymorphic DNA changes have been reported at the base pair, kilobase pair, megabase pair and/or chromosomal levels in euchromatin (non-repetitive DNA) as well as in heterochromatin (repetitive DNA). The following genetic polymorphisms, listed according to their sizes, have been classified [13]:
-
Single nucleotide polymorphisms (SNPs) (1 base pair exchanges);
-
Microsatellites (1–10 base pair repeats);
-
Small-scale insertion/inversion/deletion/duplication polymorphisms (invs/ins/indels/invdups) (1–50 base pairs in size);
-
Minisatellites (10–100 base pairs in size);
-
Small-scale repetitive elements (SSREs) (0.1–0.8 kilobase pairs in size);
-
Submicroscopic copy number variants (CNVs) (in the megabase pair range);
-
Chromosomal heteromorphisms (CHs) (in the several megabase pair range);
-
Euchromatic variants (EVs) (in the several megabase pair range).
2. Repetitive Elements in Humans
2.1. Variable Number of Tandem Repeats (VNTRs)
Microsatellites and minisatellites (defined as VNTRs) are 1 to ~10 bp and 10 to 100 bp repeats, respectively. They are mainly found in larger clusters in (peri)centromeric and (sub)telomeric regions but also dispersed along all chromosomes [1][13]. The designation ‘satellite’ originates from experiments in the 1970s: the isopycnic centrifugation of DNA led to a major peak and a few side peaks; the latter were called satellite peaks. DNA extracted therefrom was designated as satellite DNA [1]. As summarized in [1], satellite DNAs have been divided into several subgroups, as shown in Table 2. These belong to the aforementioned classes 2, 4 and 5 and are also involved in class 7: the formation of CHs.
Table 2. Satellite DNAs in humans.
| Type | Length of Basic Units (bp) | Class of Genetic Polymorphisms |
|---|---|---|
| Satellite I DNA | 17–25 | 4 |
| Satellite II DNA | 5 | 2 |
| Satellite III DNA | 5 interspersed with ~10 bp of definite sequence | 2 |
| α-satellite DNA | 171 | 5 |
| β-satellite DNA | 68–69 | 4 |
| γ-satellite DNA | 220 | 5 |
Minisatellites are 1 to ~10 bp repeats, which are species-specifically distributed along all chromosomes, and are frequently applied in the molecular cytogenetic characterization of newly discovered or cytogenetically poorly studied species [14]. In human genetics and forensics, microsatellites are analyzed with other intentions: as each human being has an (almost) unique microsatellite pattern, polymerase chain reaction-based analyses can be informative to study uniparental disomy, to perform paternity testing or to identify a perpetrator [15]. These microsatellites are still best understood as being repetitive polymorphic DNA that does not influence the phenotype (see also Section 2.2.1).
The best known microsatellite may be the 6 bp repeat of the telomeric sequence 5′-TTAGGG-3′. The preferred model system for humans, the lab mouse (Mus musculus), carries large terminal telomeric repeat blocks, which are practically not affected by aging [16]. In contrast, telomeric repeats in humans are notably degraded in somatic cells over their lifetime [17]. While it seems to be common sense that “telomeres protect chromosome ends from degradation and inappropriate DNA damage response activation through their association with specific factors” [18], their role in aging at least is under discussion [19], as no person has died yet from ‘too short telomeres’. As is typical for microsatellites, telomeric repeats are not only found at the chromosomal ends; there are also interstitial telomeric sequences (ITSs). At least some of these ITSs are interpreted as remnants from chromosome end-to-end-fusions during evolution [20]. Overall, the function of telomeric microsatellites in the cell and possibly the aging phenotype has been identified. Thus, this is the first indication that microsatellites may not only be a meaningless vestige of nature, but necessary for the biology of each living (human) cell.
Minisatellites consist of 10–100 bp repeats and are predominantly located at pericentric and subtelomeric regions but can also be found throughout the genome at thousands of different locations. Minisatellites, as in the case of microsatellites, are characterized by high mutation rates and high diversity in populations. A subgroup of minisatellites has even been shown to be hypermutable when cells are subjected to genotoxic agents [21].
2.2. Small-Scale Repetitive Elements (SSREs)
The gain, loss and insertion of DNA, which is constituted by 0.1 to 8 kb repeats, are called SSREs. Such SSREs can be slightly to highly repetitive, but the majority of them is (individually) invisible in light microscopy, as they do not reach 5 Mb in size. The latter is the lower level of resolution in banding cytogenetics [1]. Of special interest in the present context of an RNA virus–caused pandemic [22], major parts of SSREs are possibly of retroviral origin. During evolution, they may have become ‘normal’ components of eukaryotic genomes [23]. These retroviral-origin DNA repeats are predominantly grouped into ‘long interspersed nuclear elements’ (LINEs: 6–8 kb unit length) and ‘short interspersed nuclear elements’ (SINEs: 0.1–0.4 kb unit length). Furthermore, there are long terminal repeats that account for 8.3% of human genomes (0.2–3 kb unit length) [24].
-
LINEs are formed by a group of mostly truncated retrotransposons and constitute >20% of the human genome. Three types of LINEs have been identified: LINE1 (~516,000 copies), LINE2 (~315,000 copies) and LINE3 (~37,000 copies). In fact, in humans, there are ~100 active LINEs per genome, which can still amplify and integrate at new genomic sites, as they comprise reverse transcriptase [19][20][21].
-
Furthermore, SINEs derived from another subclass of retrotransposons provide ~13% of the human genome, with the feature that they can normally only become transcriptionally active if induced during infection of the human carrier by multiple DNA viruses or supported by LINE1 elements [25][26][27][28].
‘Polymorphic mitochondrial insertions’ (NumtS) are another polymorphic nuclear DNA of eukaryotes; they can be understood as a result of ongoing integration of mitochondrial DNA into the eukaryotic cell’s nucleus. More than 1000 NumtS are known in humans thus far [29]. As the mitochondrial circular genome is ~16.5kb in size, NumtS are normally shorter but can be arranged in repeats. Here, it is important to note that mitochondria are remnants of endosymbiotic organisms living in eukaryotic cells. To the best of the author’s knowledge, it is not clear whether the integration of NumtS may be, at least in part, dependent on LINE1 [30]. However, recently, we identified an exceptional case of an insertion of a cytogenetically visible NumtS block in chromosome 14 in a healthy carrier [31].
Furthermore, there is a subset of human satellite DNA (Table 2) that also belongs to the SSREs. They are DNA stretches of ~170 bp in length, can be found in low-copy repeats along the whole chromosome and are concentrated around centromeres. As detailed in [1], they form higher-order repeat (HOR) units with hundreds to tens of thousands of repeats close to the centromere. Accordingly, satellite DNAs (including classes 2 and 4) make up about 8–10% of the human genome; e.g., α-satellites are annotated at 44,058 loci covering 0.1% of the genome [32]. Interestingly, although these α- and γ-satellite sequences have been cloned, sequenced and known for decades, the majority of them are not included in genome browsers. Their sizes are variable between individuals; however, the ranges of the regions that they span have been previously reported to be between ~0.1 and 5 Mb [1]. At least some of these satellite DNAs are transcribed into RNA, but their role is yet unresolved. The fact that α-satellite DNA, for example, is expressed under cellular stress supports the idea that the alteration of heterochromatic to transcriptionally active regions could be correlated with genomic instability and oncogenesis; further supporting this notion is the fact that the tumor suppressor PKNOX1 inhibits such satellite expression. Thus, histone methylation is important for satellite DNA expression, too. As the methylation status is also altered by heat shock treatment, it is not surprising that α-satellite sequences in chromosomes 12 and 15 were proven to be expressed after a heat shock in 2004 [1].
Overall, it is also unclear if and what influence the gain or loss of such satellite-DNA-based SSRE stretches (variations may be up to several 10 Mb in size) may have on an individual. Such changes are still generally considered to have no consequences; however, this seems to be unlikely [1][33] and is controversial [33][34].
2.3. Chromosomal Heteromorphisms (CHs)
CHs are karyotypic alterations frequently found within a certain percentage of the healthy population and are clearly visible under light microscopy. CHs include the gain, loss or inversion of cytogenetically visible heterochromatic material. CHs are constituted by micro- and minisatellites, α-, β- and other satellite DNAs, often organized in HORs [1]. In humans, such heteromorphic regions are located in the (peri)centric regions of all chromosomes, at the end of the long arm of the male Y-chromosome and in the short arms of acrocentric chromosomes (chromosomes 13, 14, 15, 21 and 22). The repeat units of HORs are similar, with 95–100% identity. Within these HORs, α-satellite monomers are often intermingled with other repeats, like SINEs, LINEs, LTRs or β-satellites [35]. The hundreds of different possible human CHs are summarized elsewhere and in Table 3 [1][36]. Additionally, some examples of CHs are shown in Figure 1.
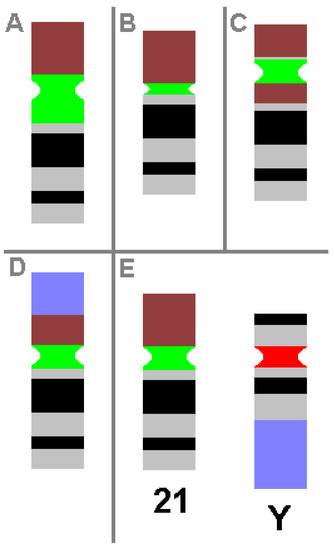
Figure 1. Schematic depiction of CHs, as known from cytogenetic diagnostics. Possible examples of centromeric amplification (A), centromeric diminution (B), pericentric inversion (C) and translocation (D) in chromosome 21 are shown, compared to a ‘normal’ chromosome 21 and a normal Y-chromosome (E). For clarity, the short arm of chromosome 21 is depicted in brown, the centromeric regions of chromosome 21 and Y-chromosome are in green and red, respectively, and the heterochromatic region of the Y-chromosome in the long arm is in blue. All aberrations shown do not cause any clinical problems.
Table 3. Number of different types of human heterochromatic chromosomal heteromorphisms (CHs) found in all chromosomes according to [36].
| Centromeric Amplification or Diminution | Pericentric Inversion | Others (e.g., Insertions or Translocations) | |
|---|---|---|---|
| Number of types found | 129 | 23 | 70 |
Even though CHs are mainly considered a cytogenetic diagnostic problem, in exceptional cases, they can be useful in terms of the following:
-
determination of paternity;
-
differentiation of mono- and dizygotic twins;
-
determination of the parental origin of derivative chromosomes or of haploid sets in polyploidy or chimera;
-
detection of maternal cell contamination in amniotic fluid cell cultures;
-
follow-up of bone marrow transplantations; or
-
identification of some genetic linkages [1].
Finally, a special CH must be added here concerning the ‘polymorphism in chromosome numbers’. This is present in many species as a supernumerary ‘B’ chromosome (B), which is nicely defined by Ahmad and Martins as “extra karyotype units in addition to A chromosomes and found in some fungi and thousands of animals and plant species. Bs are uniquely characterized due to their non-Mendelian inheritance. A classical concept based on cytogenetics and genetics is that Bs are selfish and abundant with DNA repeats and transposons, and in most cases, they do not carry any function” [37]. In humans, the existence of Bs is under discussion. Some of the so-called small supernumerary marker chromosomes (sSMCs) could be candidates for human Bs [38][39]. About 50% of sSMCs only carry heterochromatic DNA, which is also amplified in CHs.
References
- Liehr, T. Benign & Pathological Chromosomal Imbalances; Microscopic and Submicroscopic Copy Number Variations (CNVs) in Genetics and Counseling, 1st ed.; Academic Press: Berlin, Germany, 2014.
- Mkrtchyan, H.; Gross, M.; Hinreiner, S.; Polytiko, A.; Manvelyan, M.; Mrasek, K.; Kosyakova, N.; Ewers, E.; Nelle, H.; Liehr, T.; et al. The human genome puzzle—The role of copy number variation in somatic mosaicism. Curr. Genom. 2010, 11, 426–431.
- McGowan-Jordan, J.; Hastings, R.; Moore, S. (Eds.) ISCN 2020: An International System for Human Cytogenomic Nomenclature (2020); Karger: Basel, Switzerland, 2020.
- Salzberg, S.L. Open questions: How many genes do we have? BMC Biol. 2018, 16, 94.
- Statistics about the Current GENCODE Release (Version 37). Available online: https://www.gencodegenes.org/human/stats.html (accessed on 18 January 2021).
- NCBI Gene Statistics. Available online: https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Homo_sapiens/109/ (accessed on 18 January 2021).
- Human Assembly and Gene Annotation. Available online: https://www.ensembl.org/Homo_sapiens/Info/Annotation (accessed on 18 January 2021).
- GeneCards®: The Human Gene Database. Available online: https://www.genecards.org/ (accessed on 18 January 2021).
- Bessenyei, B.; Márka, M.; Urbán, L.; Zeher, M.; Semsei, I. Single nucleotide polymorphisms: Aging and diseases. Biogerontology 2004, 5, 291–303.
- Liehr, T. Human Genetics—Edition 2020: A Basic Training Package, 1st ed.; Epubli: Berlin, Germany, 2020.
- Zhao, T.; Duan, Z.; Genchev, G.Z.; Lu, H. Closing human reference genome gaps: Identifying and characterizing gap-closing sequences. G3 (Bethesda) 2020, 10, 2801–2809.
- De Bustos, A.; Cuadrado, A.; Jouve, N. Sequencing of long stretches of repetitive DNA. Sci. Rep. 2016, 6, 36665.
- Liehr, T. Repetitive elements, heteromorphisms and copy number variants. In Chromosomics, 1st ed.; Academic Press: Cambridge, MA, USA, 2021.
- Sassi, F.M.C.; Oliveira, E.A.; Bertollo, L.A.C.; Nirchio, M.; Hatanaka, T.; Marinho, M.M.F.; Moreira-Filho, O.; Aroutiounian, R.; Liehr, T.; Al-Rikabi, A.B.H.; et al. Chromosomal Evolution and Evolutionary Relationships of Lebiasina Species (Characiformes, Lebiasinidae). Int. J. Mol. Sci. 2019, 20, 2944.
- Liehr, T. Uniparental Disomy (UPD) in Clinical Genetics. A Guide for Clinicians and Patients, 1st ed.; Springer: Berlin, Germany, 2014.
- Goyns, M.H.; Lavery, W.L. Telomerase and mammalian ageing: A critical appraisal. Mech. Ageing Dev. 2000, 114, 69–77.
- Araújo Carvalho, A.C.; Tavares Mendes, M.L.; da Silva Reis, M.C.; Santos, V.S.; Tanajura, D.M.; Martins-Filho, P.R.S. Telomere length and frailty in older adults-A systematic review and meta-analysis. Ageing Res. Rev. 2019, 54, 100914.
- Ye, J.; Renault, V.M.; Jamet, K.; Gilson, E. Transcriptional outcome of telomere signalling. Nat. Rev. Genet. 2014, 15, 491–503.
- Khokhlov, A.N. Does aging need its own program, or is the program of development quite sufficient for it? Stationary cell cultures as a tool to search for anti-aging factors. Curr. Aging Sci. 2013, 6, 14–20.
- Liehr, T.; Buleu, O.; Karamysheva, T.; Bugrov, A.; Rubtsov, N. New insights into Phasmatodea chromosomes. Genes 2017, 8, 327.
- Vergnaud, G.; Denoeud, F. Minisatellites: Mutability and genome architecture. Genome Res. 2000, 10, 899–907.
- Roychoudhury, S.; Das, A.; Sengupta, P.; Dutta, S.; Roychoudhury, S.; Choudhury, A.P.; Ahmed, A.B.F.; Bhattacharjee, S.; Slama, P. Viral Pandemics of the Last Four Decades: Pathophysiology, Health Impacts and Perspectives. Int. J. Environ. Res. Public Health 2020, 17, 9411.
- Holmes, E.C. The evolution of endogenous viral elements. Cell Host Microbe 2011, 10, 368–377.
- Long Terminal Repeat. Available online: https://de.wikipedia.org/wiki/Long_Terminal_Repeat (accessed on 18 January 2021).
- Richardson, S.R.; Faulkner, G.J. Heritable L1 retrotransposition events during development: Understanding their origins: Examination of heritable, endogenous L1 retrotransposition in mice opens up exciting new questions and research directions. Bioessays 2018, 40, e1700189.
- Streva, V.A.; Jordan, V.E.; Linker, S.; Hedges, D.J.; Batzer, M.A.; Deininger, P.L. Sequencing, identification and mapping of primed L1 elements (SIMPLE) reveals significant variation in full length L1 elements between individuals. BMC Genom. 2015, 16, 220.
- Long Interspersed Nuclear Element. Available online: https://en.wikipedia.org/wiki/Long_interspersed_nuclear_element (accessed on 18 January 2021).
- Tucker, J.M.; Glaunsinger, B.A. Host noncoding retrotransposons induced by DNA viruses: A SINE of infection? J. Virol. 2017, 91, e00982-17.
- Dayama, G.; Emery, S.B.; Kidd, J.M.; Mills, R.E. The genomic landscape of polymorphic human nuclear mitochondrial insertions. Nucleic Acids Res. 2014, 42, 12640–12649.
- Hoser, S.M.; Hoffmann, A.; Meindl, A.; Gamper, M.; Fallmann, J.; Bernhart, S.H.; Müller, L.; Ploner, M.; Misslinger, M.; Kremser, L.; et al. Intronic tRNAs of mitochondrial origin regulate constitutive and alternative splicing. Genome Biol. 2020, 21, 299.
- Lutz-Bonengel, S.; Niederstätter, H.; Naue, J.; Koziel, R.; Yang, F.; Sänger, T.; Huber, G.; Berger, C.; Pflugradt, R.; Strobl, C.; et al. Evidence for multi-copy MEGA-NUMTs in the human genome. Nucleic Acids Res. 2021, gkaa1271.
- Matylla-Kulinska, K.; Tafer, H.; Weiss, A.; Schroeder, R. Functional repeat-derived RNAs often originate from retrotransposon-propagated ncRNAs. Wiley Interdiscip. Rev. RNA 2014, 5, 591–600.
- Garrido-Ramos, M.A. Satellite DNA: An evolving topic. Genes 2017, 8, 230.
- Liehr, T. Benign and pathological gain or loss of genetic material—About microscopic and submicroscopic copy number variations (CNVs) in human genetics. Tsitologiia 2016, 58, 476–477.
- Chen, J.M.; Chuzhanova, N.; Stenson, P.D.; Férec, C.; Cooper, D.N. Meta-analysis of gross insertions causing human genetic disease: Novel mutational mechanisms and the role of replication slippage. Hum. Mutat. 2005, 25, 207–221.
- Liehr, T. Cases with Heteromorphisms. 2021. Available online: http://cs-tl.de/DB/CA/HCM/0-Start.html (accessed on 18 January 2021).
- Ahmad, S.F.; Martins, C. The modern view of B chromosomes under the impact of high scale omics analyses. Cells 2019, 8, 156.
- Liehr, T. A guide for human geneticists and clinicians. In Small Supernumerary Marker Chromosomes (sSMC), 1st ed.; Springer: Berlin, Germany, 2012.
- Liehr, T. Small Supernumerary Marker Chromosomes. 2021. Available online: http://cs-tl.de/DB/CA/sSMC/0-Start.html (accessed on 18 January 2021).

