 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Federica Piancone | + 2414 word(s) | 2414 | 2021-02-24 05:10:25 | | | |
| 2 | Vivi Li | Meta information modification | 2414 | 2021-03-03 07:19:19 | | |
Video Upload Options
Neurodegenerative diseases are chronic, progressive disorders that occur in the central nervous system (CNS). They are characterized by the loss of neuronal structure and function and are associated with inflammation. Inflammation of the CNS is called neuroinflammation, which has been implicated in most neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS). Much evidence indicates that these different conditions share a common inflammatory mechanism: the activation of the inflammasome complex in peripheral monocytes and in microglia, with the consequent production of high quantities of the pro-inflammatory cytokines IL-1β and IL-18. Inflammasomes are a group of multimeric signaling complexes that include a sensor Nod-like receptor (NLR) molecule, the adaptor protein ASC, and caspase-1. The NLRP3 inflammasome is currently the best-characterized inflammasome. Multiple signals, which are potentially provided in combination and include endogenous danger signals and pathogens, trigger the formation of an active inflammasome, which, in turn, will stimulate the cleavage and the release of bioactive cytokines including IL-1β and IL-18.
1. Introduction
Neuroinflammation plays a key role in the onset and the progression of several neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), multiple sclerosis (MS), and amyotrophic lateral sclerosis (ALS) [1][2][3].
Nevertheless, it has to be considered that, in primary neurodegenerative diseases characterized by the accumulation of misfolded proteins like AD and PD, it is not clear if inflammation might be the primary cause of disease or a reaction to pathology. Indeed, the pathophysiological hypothesis of neurodegenerative diseases relies on the fact that some proteins, changing their conformations, aggregate into fibrils or oligomers, resulting in neurotoxicity and leading to neurodegeneration and inflammation [4][5][6][7]. Neuroinflammation is a physiological response to exogenous and endogenous insults that target the central nervous system (CNS) and represents a protective response in the brain, but excessive inflammatory responses are detrimental to the CNS. Several inflammation-inducing stimuli, such as damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs), are recognized by multiprotein complexes, called inflammasomes. This elicits a pro-inflammatory response mediated by the release of the inflammatory cytokines IL-1β and IL-18 [8]. The nucleotide-binding oligomerization domain leucine-rich repeat and pyrin domain-containing protein 3 (NLRP3) inflammasome, one of the most intensively investigated inflammasomes, has been reported to play a key role in neurodegenerative diseases.
Inflammasomes are a group of cytosolic multiprotein complexes that consist of a sensor molecule (NLR, AIM2-like receptors, ALR, and pyrin receptors), the adaptor apoptosis-associated speck-like protein, which contains a caspase recruitment domain (ASC), and pro-caspase-1 [9][10]. Inflammasome-inducing stimuli trigger the oligomerization of pattern recognition receptors (PRR) and the recruitment of pro-caspase-1 into the complex, leading to the generation of active caspase-1 that, consequently, will cleave inactive pro-peptides pro-IL-1β and pro-IL-18 into mature cytokines. Importantly, caspase-1 can also induce a pro-inflammatory form of cell death, pyroptosis, that features early plasma membrane rupture, thereby releasing the soluble intracellular fraction that fuels the inflammatory response [9][11]. NLRP3, as shown in Figure 1, is the best-characterized inflammasome; its activation involves a two-step process. A first signal, or “priming” signal, results in the NF-kB-dependent transcriptional upregulation of NLRP3 and pro-IL-1β, but also controls post-translational modifications of NLRP3 [12]. This initial trigger is followed by a second “activation” signal that induces the oligomerization and activation of the NLRP3 inflammasome. Besides this “canonical” NLRP3 inflammasome activation pathway, a “noncanonical” NLRP3 activatory pathway has been described. This pathway involves the activation of caspase-11 in mice (or its human orthologs caspase-4 and caspase-5) by cytosolic LPS, the induction of pyroptosis through the cleavage of gasdermin D (GSDMD), and the release of high mobility group box 1 protein (HMGB1), resulting in the production of IL-1β [9][13]. In both cases, the activation of NLRP3 inflammasome results in the cleavage of the pro-inflammatory IL-1β and IL-18; this leads to the generation of the biological active form of these proteins that initiates inflammatory signaling cascades, contributing to neuronal injury, cell death, and neuroinflammation [14][15].
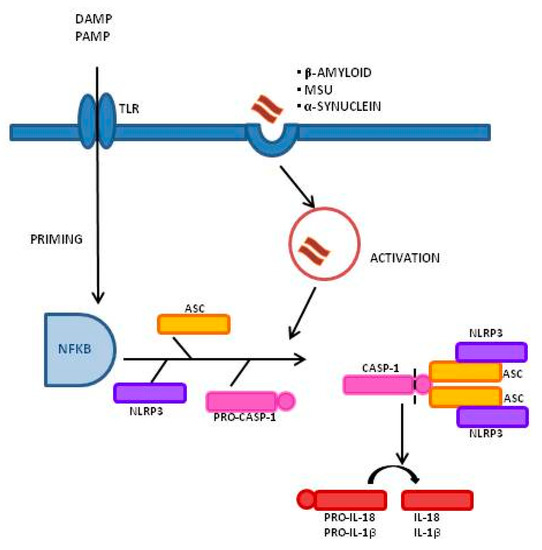
Figure 1. Inflammasome activation process and signaling mechanism: A Two-Signal Model for NLRP3 Inflammasome Activation. The priming signal (signal 1, left) is provided by damage-associated molecular patterns (DAMP) or by pathogen-associated molecular patterns (PAMP), leading to the activation of the transcription factor NF-κB and subsequent upregulation of NLRP3, pro-interleukin-1β (pro-IL-1β), and pro-interleukin-IL-18 (pro-IL-18). The activation signal (signal 2, right) is provided by a variety of stimuli including β-amyloid, α-synuclein, and Monosodium Urate Crystals (MSU), leading to the assembly and formation of the NLRP3 inflammasome through the combination of NLRP3, ASC, and procaspase-1, and leading to the production of caspase-1, which catalyzes the transformation from pro-IL-1β and pro-IL-18 into IL-1β and IL-18.
2. Alzheimer’s Disease
AD is a highly prevalent form of dementia characterized by the accumulation of extracellular amyloid beta (Aβ) plaques in the brain, neuronal cell death, and neuroinflammation. The immunological scenario of AD-associated neuroinflammation includes an increased production of pro-inflammatory cytokines, a reduced activity of Treg lymphocytes, and the dysregulation of immune-mediated mechanisms of tolerance. Activation of the NLRP3 inflammasome is strongly suggested to play a role in AD-associated neuroinflammation, as shown by results indicating that the concentration of IL-1β and IL-18 is increased in this disease [16][17][18][19].
The NLRP3 inflammasome activation by fibrillar Aβ was initially described in 2008 by Halle et al., who demonstrated that a concentration of 5 mM of fibrillar Aβ induced the production of IL-1β in a NLRP3- and ASC-dependent manner [20]. This result was confirmed by Heneka and colleagues, who showed that NLRP3-deficiency in the transgenic APP/PS1 double-transgenic mouse models of AD, which overexpressed mutated forms of the gene for human amyloid precursor protein (APP) and presenilin 1 (PS1), decreased neuroinflammation, reducing Aβ accumulation and improving neuronal function [21]. Results from other groups showed that NLRP3 or caspase-1 deletion in APP/PS1 mice promoted the differentiation of microglia to an anti-inflammatory M2 phenotype, with decreased secretion of caspase-1 and IL-1β [22]. Further support to the involvement of the NLRP3 inflammasome in the pathogenesis of AD was offered by results showing that Aβ induced the processing of pro-IL-1β into mature IL-1β in the microglia via activation of NLRP3 inflammasome [23].
The NLRP3 inflammasome may, however, not be the only inflammasome that contributes to the pathogenesis of AD [24][25]. Thus, Kaushal and colleagues showed that NLRP1 mRNA was increased in AD neurons and colocalized with caspase-6. Notably, these authors demonstrated that the NLRP1-caspase-1-caspase-6 pathway was involved in the accumulation of Aβ42 in serum-deprived neurons [26]. Additional results indicating that NLRP1 mRNA was significantly increased in sporadic and familial AD hippocampal and cortical neurons offer further support to the hypothesis that the NLRP1 inflammasome plays an important pathogenetic role in AD [26].
It is important to underline that, in addition to the inflammatory responses mediated by reactive astrocytes and by activated microglia in the CNS, an activation of the peripheral immune response is also observed and is suggested to contribute to neuroinflammation [27][28][29]. Such peripheral immune response is possibly an attempt to contrast the formation or the extension of Aβ plaques [30][31][32][33][34][35][36][37][38][39] and is associated with the stimulation of peripheral monocytes that are recruited to the CNS. Once these cells reach the CNS, though, they can contribute to the activation of the NLRP3 inflammasome [15]. To further underline the complexity of the immunological impairment that accompanies AD, recent results suggested that alterations of the microbiota might be involved in the generation of the AD-associated inflammatory milieu. Thus, a positive correlation between abundance of the inflammatory bacteria belonging to the taxon Escherichia/Shigellain in stool samples and blood levels of NLRP3 and IL-1β was observed in cognitively impaired elderly individuals with brain amyloidosis [40]. These data thus put forward the hypothesis that dysbiosis results in peripheral inflammation, which is mediated by the activation of the NLRP3 inflammasome and reverberates into the CNS.
Notably, recent in vitro studies conducted in human leukemia monocytic cell line (THP1)-derived macrophage stimulated with Aβ indicated that Leishmania infection down-regulates NLRP3 inflammasome activation, significantly reducing ASC-speck formation, thus favoring the generation of an anti-inflammatory milieu, possibly protecting against AD development [41]. This finding could explain why, despite its strong association with AD risk in industrialized populations, the Apolipoprotein E4 (ApoE4) allele was shown to be associated with improved cognitive functions in members of remote Amazonian tribes. In these populations, the E4 allele has been demonstrated to confer survival in response to infection by parasites that, in turn, could reduce inflammation by reducing the activation of NLRP3 [42].
3. Multiple Sclerosis
MS is an autoimmune demyelinating disease of the CNS characterized by immune cell infiltration from the periphery into the CNS as well as by the activation of the microglia and astrocytes, which together promote neuroinflammation and neurodegeneration [43]. A number of studies have suggested the involvement of the NLRP3 inflammasome in the pathogenesis of MS. Gris and colleagues, in 2010 [44], were the first to suggest the critical role of Nlrp3 gene in the development of experimental autoimmune encephalomyelitis (EAE), the most commonly used experimental model for human MS [45]. Results from their study showed that the absence of Nlrp3 gene resulted in diminished Th1 and Th17 encephalitogenic responses [44]. In line with this evidence, Peelen et al. reported that the expression level of the inflammasome-related genes NLRP3, IL-1β, and caspase-1, was increased in peripheral blood mononuclear cell (PBMC) from relapsing-remitting (RR) MS patients compared to healthy controls [46].
Results from other groups showed the up-regulation of caspase-1 and IL-1β proteins in PBMCs and cerebrospinal fluid (CSF) of MS patients [47][48]. Moreover, caspase-1 expression was shown to be elevated in MS plaques and PBMC of MS patients [49][50]; taken together these observations lead to the proposal of using serum caspase-1 and ASC protein concentrations as candidate biomarkers for MS onset [51]. IL-18 concentration was observed to be augmented, as well, in serum, CSF, and PBMCs of MS patients [44][52][53][54]. Furthermore, a study by de Jong et al. showed that the increase of IL-1β in CSF was concomitant with a depletion of the IL-1 receptor antagonist (IL-1Ra), an anti-inflammatory protein that antagonizes the binding of IL-1β to its receptor [55]. An indirect support to the role played by IL-1β—a prototypical NLRP3 inflammasome activation-derived cytokine—in the pathogenesis of MS stems from the observation that successful treatment of disease relapses in MS patients with glatiramer acetate or IFNβ results in the increase of endogenous IL-1Ra concentration [56][57]. Notably, IL-18 and IL-1β promote, respectively, IFNγ and IL-17 production by Th1 cells and Th17 cells, two functional T helper lymphocyte subsets that we repeatedly described to play a pivotal role in MS pathogenesis.
The canonical NLRP3 inflammasome requires caspase-1 activation for IL-1β and IL-18 processing. Recent results nevertheless indicated that T cell intrinsic inflammasome activity could drive IL-1β and IL-18 production via caspase-8 activation independently from caspase-1 activation [58][59]. Recent results reinforced a central role for the NLRP3/caspase-8 inflammasome pathway in MS by showing that stimulation of PBMCs from primary progressive MS (PPMS) patients with Monosodium Urate Crystals (MSU) resulted in a significant increase in the expression of NLRP3 and ASC-speck protein and in IL-18 and caspase-8 production. The NLRP3/caspase-8 inflammasome pathway is activated in PPMS, possibly as a consequence of hyperuricemia. Thus, levels of uric acid are upregulated in the CSF of MS patients [60], and the serum uric acid level in patients is potentially associated with susceptibility of MS [61]. Taken together, these results support the hypothesis of hyperuricemia as a common detrimental condition that characterizes MS via the activation of the NLRP3/caspase-8 inflammasome pathway [62].
Finally, the expression of P2X7R, a purinergic receptor that detects and amplifies the release of ATP and, as a consequence, the activation of NLRP3 inflammasome, was shown to be elevated in spinal cords of MS patients [63][64]. In line with this evidence, other studies have shown an association between gain-of-function single nucleotide polymorphisms in the P2X7 receptor gene and MS [65]. On the other hand, glatiramer acetate, one the immunomodulator drugs used for MS, was shown to reduce P2X7R expression [66], suggesting the contribution of extracellular ATP to the pathogenesis of MS. Taken together, these results seem to suggest that endogenous metabolic danger signals, ATP, and uric acid are likely to all be involved in the activation of the NLRP3 inflammasome pathway observed in MS.
4. Pharmacological Modulation of the Inflammasome
Given the role of the NLRP3 inflammasome in neuroinflammation, a number of studies has been conducted in the exploration of possible therapeutic pathways for neurodegenerative diseases through the inhibition of the NLRP3 inflammasome.
To date, NLRP3 inhibitors can be divided into those that directly inhibit NLRP3 or those that mediate NLRP3 inactivation as a consequence of the inhibition of inflammasome components or related signaling events. Available compounds act mainly by inhibiting the products of inflammasome activation, for example, by impeding the biological effects of IL-1β via the use of either anti-IL-1β antibodies or IL-1Ra; notably, no effective anti-IL-18 therapies are currently available [67]. Three biologics are approved by the US Food and Drug Administration (FDA) for multiple inflammatory diseases: canakinumab, an IL-1β- neutralizing antibody; anakinra, a recombinant IL-1 receptor antagonist; and rilonacept, a decoy receptor that binds IL-1β and IL-1α. Nevertheless, although their efficacy has been demonstrated for autoinflammatory diseases, there no report of their use in clinical trials in neurodegenerative diseases [68]. Because IL-1β production can be mediated by other inflammasomes, specific inhibitors that directly target the NLRP3 inflammasome could be a better option for treatment of diseases in which inflammation is the consequence of NLRP3 activation.
Some compounds have shown an inhibitory effect in vitro on NLRP3 inflammasome activation, including MCC950 [69], β-hydroxybutyrate (BHB) [70], Bay 11- 7082 [71], dimethyl sulfoxide (DMSO) [72], and type I interferon [73]. However, most of these inhibitors are relatively nonspecific and have low efficacy. Among the direct NLRP3 inhibitors, the diarylsulfonylurea compound MCC950 (originally reported as CRID3/CP-456773) is the most potent and specific for NLRP3. MCC950 demonstrated therapeutic efficacy against several preclinical immunopathological models, including EAE [69], AD [22], and PD [74]. However, this compound is currently not approved by the FDA for the therapy of neurodegenerative disease.
Using a different approach, Stavudine (d4T), an antiviral nucleoside reverse transcriptase inhibitor (NRTIs) designed to target HIV, was recently shown to down-modulate NLRP3 inflammasome activation in mice [75]. Additional data confirmed the ability of this compound to hamper NLRP3 inflammasome activation in an in vitro model of AD by reducing NLRP3 assembly as well as IL-18 and caspase-1 production and stimulating amyloid-beta autophagy by macrophages [18].
Given the lack of effective drugs in the therapy of chronic neurodegenerative conditions and the role of NLRP3 inflammasome in the pathogenesis and progression of these diseases, efforts should be made to develop effective therapeutic strategies, possibly including those targeting the NLRP3 inflammasome.
References
- Schain, M.; Kreisl, W.C. Neuroinflammation in neurodegenerative disorders-a review. Curr. Neurol. Neurosci. Rep. 2017, 17, 25.
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases. Mol. Med. Rep. 2016, 13, 3391–3396.
- Liu, Z.; Cheng, X.; Zhong, S.; Liu, C.; Liu, F.; Zhao, C. Peripheral and Central Nervous System Immune Response Crosstalk in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14, 575.
- Ciccocioppo, F.; Bologna, G.; Ercolino, E.; Pierdomenico, L.; Simeone, P.; Lanuti, P.; Pieragostino, D.; Del Boccio, P.; Marchisio, M.; Miscia, S. Neurodegenerative diseases as proteinopathies-driven immune disorders. Neural. Regen. Res. 2020, 15, 850–856.
- Bayer, T.A. Proteinopathies, a core concept for understanding and ultimately treating degenerative disorders? Eur. Neuropsychopharmacol. 2015, 25, 713–724.
- Sami, N.; Rahman, S.; Kumar, V.; Zaidi, S.; Islam, A.; Ali, S.; Ahmad, F.; Hassan, M.I. Protein aggregation, misfolding and consequential human neurodegenerative diseases. Int. J. Neurosci. 2017, 127, 1047–1057.
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci 2018, 21, 1332–1340.
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286.
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022.
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420.
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Ann. Rev. Cell. Dev. Biol. 2012, 28, 137–161.
- Yang, J.; Liu, Z.; Xiao, T.S. Post-translational regulation of inflammasomes. Cell. Mol. Immunol. 2017, 14, 65–79.
- Shi, J.Q.; Zhang, C.C.; Sun, X.L.; Cheng, X.X.; Wang, J.B.; Zhang, Y.D.; Xu, J.; Zou, H.Q. Antimalarial drug artemisinin extenuates amyloidogenesis and neuroinflammation in APPswe/PS1dE9 transgenic mice via inhibition of nuclear factor-kappaB and NLRP3 inflammasome activation. CNS. Neurosci. Ther. 2013, 19, 262–268.
- Allan, S.M. Pragmatic target discovery from novel gene to functionally defined drug target: The interleukin-1 story. Methods Mol. Med. 2005, 104, 333–346.
- Alboni, S.; Cervia, D.; Sugama, S.; Conti, B. Interleukin 18 in the CNS. J. Neuroinflamm. 2010, 7, 9.
- Awad, F.; Assrawi, E.; Jumeau, C.; Georgin-Lavialle, S.; Cobret, L.; Duquesnoy, P.; Piterboth, W.; Thomas, L.; Stankovic-Stojanovic, K.; Louvrier, C.; et al. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLoS ONE 2017, 12, e0175336.
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678.
- La Rosa, F.; Saresella, M.; Marventano, I.; Piancone, F.; Ripamonti, E.; Al-Daghri, N.; Bazzini, C.; Zoia, C.P.; Conti, E.; Ferrarese, C.; et al. Stavudine Reduces NLRP3 Inflammasome Activation and Modulates Amyloid-β Autophagy. J. Alzheimers Dis. 2019, 72, 401–412.
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 1–14.
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865.
- Heneka, M.T. Inflammasome activation and innate immunity in Alzheimer’s disease. Brain Pathol. 2017, 27, 220–222.
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O’Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316.
- Parajuli, B.; Sonobe, Y.; Horiuchi, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Oligomeric amyloid β induces IL-1β processing via production of ROS: Implication in Alzheimer’s disease. Cell Death Dis. 2013, 4, e975.
- Pontillo, A.; Catamo, E.; Arosio, B.; Mari, D.; Crovella, S. NALP1/NLRP1 genetic variants are associated with Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2012, 26, 277–281.
- Tan, M.S.; Tan, L.; Jiang, T.; Zhu, X.C.; Wang, H.F.; Jia, C.D.; Yu, J.T. Amyloid-beta induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1382.
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.; Hyman, B.; Ghetti, B.; Koller, B.H.; LeBlanc, A.C. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015, 22, 1676–1686.
- Rezai-Zadeh, K.; Gate, D.; Gowing, G.; Town, T. How to get from here to there: Macrophage recruitment in Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 156–163.
- Nascimento, C.M.; Pereira, J.R.; de Andrade, L.P.; Garuffi, M.; Talib, L.L.; Forlenza, O.V.; Cancela, J.M.; Cominetti, M.R.; Stella, F. Physical exercise in MCI elderly promotes reduction of pro-inflammatory cytokines and improvements on cognition and BDNF peripheral levels. Curr. Alzheimer Res. 2014, 11, 799–805.
- Dionisio-Santos, D.A.; Olschowka, J.A.; O’Banion, M.K. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 74.
- Town, T.; Tan, J.; Mullan, M. CD40 signaling and Alzheimer’s disease pathogenesis. Neurochem. Int. 2001, 39, 371–380.
- Town, T.; Nikolic, V.; Tan, J. The microglial “activation” continuum: From innate to adaptive responses. J. Neuroinflamm. 2005, 2, 24.
- Town, T.; Laouar, Y.; Pittenger, C.; Mori, T.; Szekely, C.A.; Tan, J.; Duman, R.S.; Flavell, R.A. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat. Med. 2008, 14, 681–687.
- Hawkes, C.A.; McLaurin, J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 1261–1266.
- Tan, M.S.; Yu, J.T.; Jiang, T.; Zhu, X.C.; Tan, L. The NLRP3 inflammasome in Alzheimer’s disease. Mol. Neurobiol. 2013, 48, 875–882.
- Townsend, K.P.; Town, T.; Mori, T.; Lue, L.F.; Shytle, D.; Sanberg, P.R.; Morgan, D.; Fernandez, F.; Flavell, R.A.; Tan, J. CD40 signaling regulates innate and adaptive activation of microglia in response to amyloid beta-peptide. Eur. J. Immunol. 2005, 35, 901–910.
- Feng, Y.; Li, L.; Sun, X.H. Monocytes and Alzheimer’s disease. Neurosci. Bull. 2011, 2, 115–122.
- Fiala, M.; Lin, J.; Ringman, J.; Kermani-Arab, V.; Tsao, G.; Patel, A.; Lossinsky, A.S.; Graves, M.C.; Gustavson, A.; Sayre, J.; et al. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer’s disease patients. J. Alzheimers Dis. 2005, 3, 221–232.
- Simard, A.R.; Rivest, S. Neuroprotective properties of the innate immune system and bone marrow stem cells in Alzheimer’s disease. Mol. Psychiatry 2006, 11, 327–335.
- Saresella, M.; Marventano, I.; Calabrese, E.; Piancone, F.; Rainone, V.; Gatti, A.; Alberoni, M.; Nemni, R.; Clerici, M. A complex proinflammatory role for peripheral monocytes in Alzheimer’s disease. J. Alzheimers Dis. 2014, 38, 403–413.
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging. 2017, 49, 60–68.
- Saresella, M.; Basilico, N.; Marventano, I.; Perego, F.; La Rosa, F.; Piancone, F.; Taramelli, D.; Banks, H.; Clerici, M. Leishmania infantum infection reduces the amyloid β42-stimulated NLRP3 inflammasome activation. Brain Behav. Immun. 2020, 88, 597–605.
- Trumble, B.C.; Stieglitz, J.; Blackwell, A.D.; Allayee, H.; Beheim, B.; Finch, C.E.; Gurven, M.; Kaplan, H. Apolipoprotein E4 is associated with improved cognitive function in Amazonian forager-horticulturalists with a high parasite burden. FASEB J. 2017, 31, 1508–1515.
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.W. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768.
- Gris, D.; Ye, Z.; Iocca, H.A.; Wen, H.; Craven, R.R.; Gris, P.; Huang, M.; Schneider, M.; Miller, S.D.; Ting, J.P. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J. Immunol. 2010, 185, 974–981.
- Ransohoff, R.M. Animal models of multiple sclerosis: The good, the bad and the bottom line. Nat. Neurosci. 2012, 15, 1074–1077.
- Peelen, E.; Damoiseaux, J.; Muris, A.H.; Knippenberg, S.; Smolders, J.; Hupperts, R.; Thewissen, M. Increased inflammasome related gene expression profile in PBMC may facilitate T helper 17 cell induction in multiple sclerosis. Mol. Immunol. 2015, 63, 521–529.
- Inoue, M.; Shinohara, M.L. NLRP3 Inflammasome and MS/EAE. Autoimmune Dis. 2013, 2013, 859145.
- Mamik, M.K.; Power, C. Inflammasomes in neurological diseases: Emerging pathogenic and therapeutic concepts. Brain 2017, 140, 2273–2285.
- Furlan, R.; Filippi, M.; Bergami, A.; Rocca, M.A.; Martinelli, V.; Poliani, P.L.; Grimaldi, L.M.; Desina, G.; Comi, G.; Martino, G. Peripheral levels of caspase-1 mRNA correlate with disease activity in patients with multiple sclerosis; a preliminary study. J. Neurol. Neurosurg. Psychiatry 1999, 67, 785–788.
- Ming, X.; Li, W.; Maeda, Y.; Blumberg, B.; Raval, S.; Cook, S.D.; Dowling, P.C. Caspase-1 expression in multiple sclerosis plaques and cultured glial cells. J. Neurol. Sci. 2002, 197, 9–18.
- Keane, R.W.; Dietrich, W.D.; de Rivero Vaccari, J.P. Inflammasome Proteins As Biomarkers of Multiple Sclerosis. Front. Neurol. 2018, 9, 135.
- Losy, J.; Niezgoda, A. IL-18 in patients with multiple sclerosis. Acta Neurol. Scand. 2001, 104, 171–173.
- Nicoletti, F.; Di Marco, R.; Mangano, K.; Patti, F.; Reggio, E.; Nicoletti, A.; Bendtzen, K.; Reggio, A. Increased serum levels of interleukin-18 in patients with multiple sclerosis. Neurology 2001, 57, 342–344.
- Chen, Y.C.; Chen, S.D.; Miao, L.; Liu, Z.G.; Li, W.; Zhao, Z.X.; Sun, X.J.; Jiang, G.X.; Cheng, Q. Serum levels of interleukin (IL)-18, IL-23 and IL-17 in Chinese patients with multiple sclerosis. J. Neuroimmunol. 2012, 243, 56–60.
- De Jong, B.A.; Huizinga, T.W.; Bollen, E.L.; Uitdehaag, B.M.; Bosma, G.P.; van Buchem, M.A.; Remarque, E.J.; Burgmans, A.C.; Kalkers, N.F.; Polman, C.H.; et al. Production of IL-1beta and IL-1Ra as risk factors for susceptibility and progression of relapse-onset multiple sclerosis. J. Neuroimmunol. 2002, 126, 172–179.
- Burger, D.; Molnarfi, N.; Weber, M.S.; Brandt, K.J.; Benkhoucha, M.; Gruaz, L.; Chofflon, M.; Zamvil, S.S.; Lalive, P.H. Glatiramer acetate increases IL-1 receptor antagonist but decreases T cell-induced IL-1β in human monocytes and multiple sclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 4355–4359.
- Nicoletti, F.; Patti, F.; DiMarco, R.; Zaccone, P.; Nicoletti, A.; Meroni, P.; Reggio, A. Circulating serum levels of IL-1ra in patients with relapsing remitting multiple sclerosis are normal during remission phases but significantly increased either during exacerbations or in response to IFN-β treatment. Cytokine 1996, 8, 395–400.
- Martin, B.N.; Wang, C.; Zhang, C.J.; Kang, Z.; Gulen, M.F.; Zepp, J.A.; Zhao, J.; Bian, G.; Do, J.S.; Min, B.; et al. T cell-intrinsic ASC critically promotes T(H)17-mediated experimental autoimmune encephalomyelitis. Nat. Immunol. 2016, 17, 583–592.
- Pierini, R.; Perret, M.; Djebali, S.; Juruj, C.; Michallet, M.C.; Förster, I.; Marvel, J.; Walzer, T.; Henry, T. ASC controls IFN-γ levels in an IL-18-dependent manner in caspase-1-deficient mice infected with Francisella novicida. J. Immunol. 2013, 191, 3847–3857.
- Amorini, A.M.; Petzold, A.; Tavazzi, B.; Eikelenboom, J.; Keir, G.; Belli, A.; Giovannoni, G.; Di Pietro, V.; Polman, C.; D’Urso, S.; et al. Increase of uric acid and purine compounds in biological fluids of multiple sclerosis patients. Clin. Biochem. 2009, 42, 1001–1006.
- Liu, B.; Shen, Y.; Xiao, K.; Tang, Y.; Cen, L.; Wei, J. Serum uric acid levels in patients with multiple sclerosis: A meta-analysis. Neurol. Res. 2012, 34, 163–171.
- Piancone, F.; Saresella, M.; Marventano, I.; La Rosa, F.; Santangelo, M.A.; Caputo, D.; Mendozzi, L.; Rovaris, M.; Clerici, M. Monosodium Urate Crystals Activate the Inflammasome in Primary Progressive Multiple Sclerosis. Front. Immunol. 2018, 9, 983.
- Yiangou, Y.; Facer, P.; Durrenberger, P.; Chessell, I.P.; Naylor, A.; Bountra, C.; Banati, R.R.; Anand, P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006, 6, 12.
- Matute, C. Interaction between glutamate signalling and immune attack in damaging oligodendrocytes. Neuron. Glia. Biol. 2007, 3, 281–285.
- Oyanguren-Desez, O.; Rodriguez-Antiguedad, A.; Villoslada, P.; Domercq, M.; Alberdi, E.; Matute, C. Gain-of-function of P2X7 receptor gene variants in multiple sclerosis. Cell Calcium. 2011, 50, 468–472.
- Caragnano, M.; Tortorella, P.; Bergami, A.; Ruggieri, M.; Livrea, P.; Specchio, L.M.; Martino, G.; Trojano, M.; Furlan, R.; Avolio, C. Monocytes P2X7 purinergic receptor is modulated byglatiramer acetate in multiple sclerosis. J. Neuroimmunol. 2012, 245, 93–97.
- Netea, M.G.; van de Veerdonk, F.L.; van der Meer, J.W.; Dinarello, C.A.; Joosten, L.A. Inflammasome-independent regulation of IL-1-family cytokines. Annu. Rev. Immunol. 2015, 33, 49–77.
- Duan, Y.; Kelley, N.; He, Y. Role of the NLRP3 inflammasome in neurodegenerative diseases and therapeutic implications. Neural Regen Res. 2020, 15, 1249–1250.
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255.
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269.
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 2010, 285, 9792–9802.
- Ahn, H.; Kim, J.; Jeung, E.B.; Lee, G.S. Dimethyl sulfoxide inhibits NLRP3 inflammasome activation. Immunobiology 2014, 219, 315–322.
- Inoue, M.; Williams, K.L.; Oliver, T.; Vandenabeele, P.; Rajan, J.V.; Miao, E.A.; Shinohara, M.L. Interferon-β therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci. Signal. 2012, 5, ra38.
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066.
- Fowler, B.J.; Gelfand, B.D.; Kim, Y.; Kerur, N.; Tarallo, V.; Hiran, Y.; Amarnath, S.; Fowler, D.H.; Radwan, M.; Young, M.T. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 2014, 346, 1000–1003.

