 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Valerie Carpenter | + 5503 word(s) | 5503 | 2021-02-20 03:31:21 | | | |
| 2 | Bruce Ren | -21 word(s) | 5482 | 2021-03-02 03:21:32 | | | | |
| 3 | Bruce Ren | -21 word(s) | 5482 | 2021-03-02 03:41:37 | | |
Video Upload Options
Senolytics represent a group of mechanistically diverse drugs that can eliminate senescent cells, both in tumors and in several aging-related pathologies. Consequently, senolytic use has been proposed as a potential adjuvant approach to improve the response to senescence-inducing conventional and targeted cancer therapies. However, the translation of senolytics to the clinic faces many challenges that need to be addressed by the research community.
1. Introduction
Within the last few years, senescence has been increasingly recognized as a central component of tumor biology and the response to anti-cancer therapies. In its simplest form, senescence is a stress response that occurs subsequent to replicative-, oxidative-, oncogene-, and therapy-induced insults [1][2][3][4]. Senescent cells undergo a prolonged growth arrest, yet remain metabolically viable, and can be identified by an array of phenotypes including structural changes [5][6], increased lysosomal biogenesis (senescence-associated beta galactosidase, SA-β-gal) [7][8], dysregulated metabolism [9][10], epigenetic changes [11], and extensive alterations in gene expression [12]. Senescence is also almost universally accompanied by the secretion of various soluble and insoluble factors, termed the senescence associated secretory phenotype (SASP) [13][14]. However, despite these hallmark features, it is important to understand that the senescent phenotype can be highly variable, based on cell type and senescence-inducing stimulus. For example, Hernandez-Segura et al. revealed a heterogeneity in senescence-associated gene expression in fibroblasts depending on whether senescence was induced by replicative exhaustion, oncogene hyperactivation, or ionizing radiation . This same study also showed that the expression of key senescence-related genes was dynamic and could vary temporally [15]. Furthermore, the degradation of Lamin B1 is an established feature of senescence [16]; however, in oxidation-induced senescence, Lamin B1 may accumulate [17], providing another example of the phenotypic variability of senescence. The SASP is also heterogeneous, and its diverse composition based on cell type and stimuli has been well characterized by Basisty et al. [18]. Importantly, not all senescence-inducing stimuli will elicit a SASP; this is particularly the case for non-DNA damaging insults [19]. Finally, single cell sequencing of senescent cells has also revealed that this heterogeneity in gene expression can exist even within a monoclonal population of cells [20][21].
Premalignant and malignant (tumor) cells, although typically undergoing rapid replication, can also enter a senescent cell state, characterized by a stable growth arrest and the presence of multiple senescence hallmarks. During malignant transformation, for example, senescence can serve to delay or subvert the progression to tumorigenesis of premalignant cells, thereby acting as a tumor suppressive mechanism [22][23]. In established malignancies undergoing treatment, a plethora of anti-cancer drugs with variable mechanisms of action have been shown to promote a form of therapy-induced senescence (TIS) both in vitro and in vivo [24]. For example, conventional therapies such as etoposide, doxorubicin, and cisplatin are established inducers of TIS [25][26][27][28][29][30]. Targeted therapies, such as the BRAF inhibitor vemurafenib, EGFR inhibitor osimertinib, and hormonal therapies, including fulvestrant and androgen deprivation, have also been shown to result in senescence induction [31][32][33][34][35][36][37][38]. For an extensive review of drug classes that have been shown to promote TIS, we refer the reader to Saleh et al.. Furthermore, human tumor samples from patients treated with genotoxic therapies have demonstrated that this response is also likely conserved clinically, although the evidence in support of this premise is more limited [39][40]. As such, it is highly probable that senescent tumor cells accumulate over time, both spontaneously and as a consequence of therapy.
While TIS has long been established in the cancer field, a full understanding of how senescence may impact patient outcome has been evolving and is far from complete. The long-held traditional paradigm argued that senescence was an irreversible cell fate [41], and as such, TIS was purported to be a beneficial outcome of therapy in that it could lead to permanent abrogation of established tumor growth [42][43]. However, in recent years, multiple reports have been generated in support of the premise that cells that have entered into TIS can, in fact, escape the senescent growth arrest; this has been shown following various therapies, including topoisomerase poisons [44][45], alkylating agents [46], ionizing radiation [47][48], cyclin-dependent kinase 4/6 inhibitors [49], PARP inhibitors [50], and androgen deprivation. Furthermore, tumor cells that escape senescence have been reported to develop more aggressive phenotypes associated with increased stemness and drug resistance [51][52], although this is not universally the case, as some senescence escapers are only as neoplastic as their parental cells [53][54]. In addition, TIS in non-tumor cells has been linked with several untoward effects of cancer therapy, including cancer relapse [55], and more importantly, senescent tumor cells themselves have been demonstrated to directly account for the emergence of recurrent cancer phenotypes [56]. Therefore, while the senescent growth arrest may confer short-term advantages with regards to tumor progression, these beneficial effects may only be short-lived, and may be permissive for the development of more pernicious cancer phenotypes over an extended period of time.
The SASP is also double-edged. On the one hand, the SASP may activate the immune system and lead to tumor clearance [57]; on the other hand, the SASP can be immunosuppressive and may promote growth, epithelial-to-mesenchymal transition, and increased stemness in neighboring cells [58]. Consistent with these opposing functions, there is ample evidence showing that senescence can both contribute to deleterious effects of therapy, and conversely, be associated with better patient outcomes. These conflicting observations are likely explained by the heterogeneity of the senescent phenotype and the context in which it occurs (i.e., tumor type and treatment), emphasizing the critical need for a more in-depth understanding of TIS in the context of patient response to these therapeutic interventions [59].
In response to the identification of the potentially deleterious consequences of senescence, the development and characterization of small molecules that can modulate senescence has rapidly expanded. Senolytics, drugs that preferentially target senescent cells for cell death, were originally identified in studies of aging [60]. Of these agents, studies utilizing the BCL-XL/BCL-2 inhibitor navitoclax (ABT-263) or the dasatinib+quercetin (D+Q) combination have generated the most promising experimental data for the elimination of senescent cells. It is important to note, especially for compounds that influence multiple signaling pathways such as D+Q, the difference between senolytic and senomorphic activity. To be considered a senolytic, a compound must be capable of killing senescent cells. Alternatively, alteration of senescent phenotypes, such as the SASP, or reducing senescence induction without cell killing is more indicative of a senomorphic activity. Both ABT-263 and D+Q, for example, have been previously established to be senolytic, rather than senomorphic [61][62]. While senomorphics have been postulated to target the more unfavorable aspects of senescence, such as the inflammatory SASP [63], the use of senolytics has garnered more attention in the cancer field. These agents have now been favored by the cancer community largely under the premise that the combination—or “two-hit” approach—of senescence-inducing chemotherapies followed by senolytics may increase tumor cell killing and/or eliminate residual disease [64][65][66].
While the adoption of senolytics as adjuvant cancer therapeutics carries potential promise, multiple reports examining the effectiveness of different senolytic agents in combination with senescence-inducing therapies in cancer models have raised several issues and potential concerns. These include the lack of universality of senolytic action against different therapy-induced senescence models, the potential for systemic toxicity such as neutropenia and thrombocytopenia (in the case of ABT-263), the likelihood that resistance to the senolytics could develop, and their specificity for “harmful” senescent cells. In this review, we attempt to provide a critical assessment of current evidence in support of the utilization of senolytic therapy in cancer, some reservations relating to their clinical applicability, as well as to offer insight into how the senolytic strategy for cancer therapy might be optimized. We also propose that senolytics could be utilized initially for the purposes of reducing the likelihood of cancer recurrence based on the premise that their administration could promote the removal of senescent tumor cells that reflect a dormant population with the capacity for re-emergence and repopulation [67]. However, actually evaluating this possibility in the clinic would be extremely challenging, requiring extensive and lengthy monitoring periods, since tumor dormancy and disease recurrence is extremely variable and can occur over periods of months and possibly years. Consequently, this approach would likely be initiated, at first, in the types of cancer where current standards of care remain inadequate and which are associated with short patient-free survival periods.
2. Senolytic Therapies: Have We Hit Gold or Pyrite?
2.1. Established Success of Senolytic Therapy in the Mitigation of Aging-Associated Disease
The first step in the development of senolytics was the identification of transcriptomic signatures unique to irradiated, senescent human fat cell progenitors, or pre-adipocytes [60]. These cells showed a differential increase in pro-survival or anti-apoptotic pathways, consistent with the prolonged survival and persistence of senescent cells. Consequently, the targeted siRNA blockade of molecular targets involved in these pathways, including PI3K, p21Cip1, BCL-XL, or PAI-2, resulted in the killing of senescent cells but not their proliferating or quiescent counterparts. This paved the way for the development of small molecule inhibitors targeting essential survival pathways in senescent cells. Via the screening of 46 potential agents, the first efficacious senolytic strategy utilized the combination of dasatinib+quercetin (D+Q) . Dasatinib is a tyrosine kinase inhibitor that interferes with ephrin-dependent survival, whereas quercetin is a flavanol that inhibits various kinases, including PI3K. The D+Q combination has been shown to clear senescent cells in vitro and in vivo in a variety of senescence or aging-related models including chronic atherosclerotic vascular disease [68][69], radiation- or bleomycin-induced lung fibrosis [70], aging-associated hepatic steatosis [71], arteriovenous fistulation in chronic kidney disease [72], Alzheimer’s disease [73][74], hyperoxia-induced airway dysfunction [75], obesity-associated anxiety [76], obesity-induced metabolic dysfunction [77], osteoarthritis [78], radiation-induced bone degeneration [79] and radiation-induced ulceration [80]. D+Q has been shown to ameliorate various organ system dysfunctions associated with these disorders, improve physical dysfunction and increase survival of naturally or experimentally-induced aged mice [81]. Moreover, D+Q is being investigated in clinical trials targeting chronic kidney disease, pulmonary fibrosis and Alzheimer’s disease [NCT02848131, NCT02874989, NCT04210986]. Early results have shown that D+Q can reduce senescent cell burden in vivo [82]. Moreover, D+Q improved physical function in 13 patients with idiopathic lung fibrosis despite unremarkable changes in senescence-associated biomarkers [83].
Another frequently utilized senolytic is the BH3 mimetic, ABT-263 (navitoclax). ABT-263 was first tested as a senolytic based on the dependence of some senescent cells on the pro-survival proteins of the BCL-2 family [84]. As is the case for other BH3 mimetics, ABT-263 promotes apoptosis by binding pro-survival BCL-2 family members and preventing their association with pro-death BCL-2 family members [85]. The functional activities of BCL-2, BCL-XL, and BCL-W, but not MCL-1, are inhibited by ABT-263 [85]. Originally conceived as a cancer therapeutic, ABT-263 was synthesized using structure-based drug design and was later found to selectively induce apoptosis in senescent bone marrow hematopoietic stem cells, which resulted in bone marrow recovery following total body irradiation in mice. Notably, the senolytic action of ABT-263 appears to depend on inhibition of BCL-XL and/or BCL-W, while BCL-2 inhibition is dispensable [86]. Similar to D+Q, ABT-263 was successful in eliminating senescent cell populations in several disease models including aging-associated bone loss [87], radiation-induced lung fibrosis [88], lung emphysema [89], uterine leiomyoma [90], tau-dependent neurodegenerative disease [91], radiation-induced neurodegeneration [92], myocardial infarction (including ischemia-reperfusion injury) [93][94], heart failure [95], pulmonary hypertension [96], insulin resistance [97], osteoarthritis [98][99], synthetic implant-mediated fibrosis [100], and Duchenne muscular dystrophy [101]. ABT-263 is currently a cornerstone in preclinical studies of senolysis and remains a promising drug for use against both liquid and solid tumors [102][103][104].
Both dasatinib and navitoclax are synthetic small molecules that were initially developed as cancer therapeutics but are now under consideration for the mitigation of aging-related pathologies. Other naturally occurring substances with potential senolytic activity and low overall toxicity are also under investigation. In addition to quercetin, fisetin has also shown senolytic activity. Fisetin belongs to the flavonoid family, which are a group of natural chemicals widely available in plants, including several vegetables and fruits. Of 10 flavonoids tested, fisetin showed the greatest efficacy in eliminating subpopulations of senescent mouse and human adipose cells; fisetin increased both the median and maximum lifespan of mice, accompanied by a significant reduction of senescence-associated markers [105]. Fisetin also promoted the killing of senescent human umbilical vein endothelial cells (HUVECs) but not senescent IMR90 human lung fibroblasts or senescent primary human preadipocytes, reflecting a somewhat narrow-spectrum and cell type-specific activity [106]. Piperlongumine is a natural product found in the fruit of Piper longum that was also identified, through drug screening, to exert senolytic activity in radiation-, replicative- and oncogene-induced senescent WI-38 fibroblasts [107]. Both curcumin, a primary component in turmeric, and its synthetic analogue EF24, exhibit senolytic activity [108][109]. A number of additional compounds have been found to exhibit a range of senolytic effects, with the majority being established drugs with newly discovered ability to kill senescent cells and contribute to the amelioration of aging-related pathologies (reviewed in [24]).
2.2. The Dual Faces of Therapy-Induced Senescence
A fundamental premise underlying the use of senolytic agents as adjuvant to cancer therapy is that the accumulation of senescent tumor cells following exposure to conventional or targeted chemotherapeutics is deleterious. Since the first report to demonstrate TIS [110], it was anticipated that the induction of senescence in tumor cells would impose an irreversible growth arrest, thereby indefinitely suppressing tumor progression [111][112], supporting the development of agents that are pro-senescent. However, it has subsequently been recognized that the senescent phenotype extends beyond the induction of growth arrest and encompasses functional traits that can alter the tumor microenvironment and tumor immunosurveillance. Furthermore, the long-standing paradigm that senescence is an “irreversible” form of growth arrest has now been challenged by a growing collection of literature suggesting otherwise [113]. Our current understanding of the contribution(s) of senescence to cancer therapeutic outcomes are largely summarized below.
2.2.1. Stability of the Senescent Growth Arrest
First, recent studies have provided compelling evidence that senescent tumor cells are in a “durable” but not permanent state of growth arrest, and that a subpopulation of tumor cells can and will ultimately recover proliferative capacity. Early evidence in support of this was provided in studies by Elmore et al., wherein the evolution of senescence-resistant clones of MCF-7 breast tumor cells after exposure to Adriamycin suggested that the senescent growth arrest can potentially be evaded [114]. Then, Roberson et al. demonstrated the escape from TIS in H1299 non-small lung cancer cell model which lacks p53 and p16INK4a functions. It was subsequently shown that a variety of tumor cells exposed to genotoxic chemotherapy or ionizing radiation, with or without PARP inhibition (including lung, breast and colon), can escape from an established senescence phenotype, and resume growth, accompanied by partial or complete resolution of SA-β-gal staining and decreased expression of CDKIs (e.g., p21Cip1) and SASP factors (e.g., IL-1, IL-6, IL-8). To obviate concerns that recovery may be from non-senescent populations, work from our lab has directly observed the spontaneous division of senescent cells via live-cell imaging and high speed live cell interferometry [115]. Flow cytometry-based enrichment of SA-β-gal-positive (TIS) tumor cells implanted in immune-competent or immune-deficient animals further showed that senescent tumor cells can form viable tumors. Similar results were obtained in a model of acute myeloid leukemia (AML), in which cytosine arabinoside promoted an escapable form of senescence in AML cells. Enrichment of these senescent cells and subsequent engraftment into mice was permissive for in vivo leukemia repopulation, suggesting that the recovery of senescent tumor cells may be sufficient for recurrent disease.
While the precise mechanism for escape from TIS is likely dependent on tumor type and therapy, a few mechanisms have been described thus far. For example, in p53-null lung cancers, overexpression of Cdk-2 was sufficient to bypass TIS growth arrest. Downregulation of cell-membrane receptors for senescence-reinforcing proteins that are secreted during senescence may also be permissive for escape [116]. Finally, the ability to revert from senescence may also be attributed, at least in part, to large-scale gene expression alterations that allow for acquisition of stem cell-like characteristics, e.g., by activating the WNT signaling cascade [117]. This simultaneously allows for both proliferative recovery and the development of aggressive phenotypes (e.g., advanced migratory and invasive features). While the TIS response in tumor cells can be highly heterogenous [118], and although the ability of senescent cells to escape the growth arrest in patient tumors is still unknown, these cell-autonomous features of TIS support the strategies of eliminating senescent tumor cells using senolytics [64].
2.2.2. Impacts on the Immune System
An additional, and rather severe, limitation to our understanding of how senescence influences the efficacy of therapy and patient outcomes relates to the interaction(s) between senescent tumor cells and the immune system. The ability of senescent cells to recruit immune cells appears to be largely non-cell autonomous and mediated through the SASP. It is important to reiterate, however, that the SASP is highly heterogeneous, in part, related to the nature of the senescence-inducing stimuli and cell type. Nevertheless, there are several factors that seem to be generally common, including the pro-inflammatory factors IL-6, IL-8, IL-1Β, and GROα [119]. Activation of the SASP has been directly linked to immune-mediated senescent cell clearance. For example, endometrial stromal cells that enter senescence during decidualization secrete IL-15 and are recognized and killed by uterine natural killer (NK) cells [120]. Liver cells that undergo oncogene-mediated senescence are recognized and eliminated by CD4+ T-cells [121], and senescent hepatic stellate cells have been shown to regulate and enhance activity of M1 macrophages [122]. Recruitment of immune cells by the SASP can also create further therapeutic vulnerabilities to immune checkpoint blockade [123]. This was shown by Ruscetti et al. in a model of pancreatic cancer, where intratumoral accumulation of CD8+ T-cells following TIS conferred increased response to anti-PD-1 therapy [123]. Importantly, the milieu secreted by senescent cells, and therefore the subsequent effect on immune cells, can be modulated by p53 status [124][125], likely due to its role in establishing the senescent response. For example, in the study on hepatic stellate cells, p53 -activated cells entered senescence and showed elevated secretion of IL-6, ICAM-1, and IFNγ. p53-suppressed, proliferating cells showed elevated secretion of IL-3, IL-4, and IL-5. These profiles directly affected macrophage phenotype, with the former promoting M1 polarization and the latter favoring M2 [122]. Furthermore, murine liver carcinomas with restored p53 activity entered senescence, and were suppressed through increased inflammatory cytokine secretion and activation of NK cells, macrophages, and neutrophils [125]. Given these observations, it appears that senescence and the SASP likely favor immune system recognition and tumor cell killing, so long as senescent cells can be recognized.
It is intuitive that senescent cell accumulation during the natural aging process must be associated with the ability to escape immune surveillance. In elderly individuals, this may be due, at least in part, to the natural decline in the immune system, called immunosenescence [126]. During immunosenescence, immune cells display many of the canonical features of senescence, including elevated expression of cyclin dependent kinase inhibitors, increased SA-β-Gal, and development of the SASP [126]. Notably, activity and production of cytotoxic molecules also decreases [126]. This phenomenon has repeatedly been linked to the higher occurrence of malignancies (and other immune-regulated illnesses) in the elderly [127], and arguably, would allow for the persistence of senescent cells, both normal and cancerous. Further, tumor induced T-cell senescence has also been reported [128][129][130], and this has been shown to be an additional mechanism of immune-evasion. This may also impair T-cell activity against senescent tumor cells. Finally, there have been reports of direct suppression and evasion of otherwise-healthy (i.e., non-senescent) immune cells by senescent tumor cells. For example, in a mouse model of aging, senescent stromal cells showed enhanced secretion of IL-6 as part of their SASP, and this resulted in increased levels of suppressive myeloid cells and the creation of a tumor-permissive microenvironment [131]. PTEN-deficient, senescent prostate cancer cells can also escape detection by secreting immunosuppressive factors, although this can be reversed by inhibiting STAT3/JAK signaling [132]. Senescent dermal fibroblasts have been shown to upregulate HLA-E, which interacts with an inhibitory receptor on NK cells and cytotoxic CD8+ T-cells [133]. High expression of matrix metalloproteases (often components of the SASP) can also cleave ligands for NKG2D receptors, allowing senescent cells to “shed” the signals that would otherwise mark the cells for NK-cell killing [134]. As such, whether or not the immune system efficiently recognizes TIS cancer cells is unclear and likely contextual. Overall, however, these observations suggest that at least a subpopulation of senescent tumor cells may eventually escape immunosurveillance, which could facilitate tumor recurrence.
2.2.3. TIS in Non-Malignant Cells
It is further necessary to recognize that senescence induction in response to cancer therapy is not limited to tumor cells, as non-malignant cells can and do undergo senescence in response to chemotherapy or radiation. In fact, the induction of TIS in non-malignant cells has been linked to organ dysfunction involving the heart, kidneys, bone, bone marrow and nervous system, which all contribute to and/or may be central for many adverse effects of cancer therapy [135]. The persistence of these cells for prolonged periods, and their secretory ability, can drive an insidious inflammatory response that alters the homeostatic microenvironment [136]. Furthermore, as indicated in a previous section, the selective removal of these senescent cells, genetically or pharmacologically, was shown to result in beneficial outcomes. This was clearly demonstrated by the contribution of senescent cells accumulation to radiation-induced pulmonary fibrosis or radiation-induced skin ulceration, both common complications of the use of radiotherapy for cancer.
2.2.4. Evidence Supporting the Removal of Non-Malignant Therapy-Induced Senescent Cells
The cumulative evidence for deleterious consequences of TIS suggests that eliminating senescent cells is likely to prove beneficial, although, as indicated below, the clinical feasibility of this strategy remains to be unequivocally established. In 2017, Demaria et al. reported that the clearance of non-malignant senescent cells following doxorubicin treatment, in studies utilizing either a transgenic (p16-3MR) mouse model or the Bcl-2 family-targeting agent, navitoclax, resulted in less severe bone-marrow suppression, a decrease in cardiac dysfunction, a reduction in the development of metastases, and interference with tumor recurrence. Another study has shown that elimination of senescent-like dorsal root ganglia neurons by navitoclax alleviated both mechanical and thermal hypersensitivity in a mouse model of cisplatin-induced peripheral neuropathy [137]. Similarly, using the INK-ATTAC model, doxorubicin-induced trabecular and cortical bone loss was alleviated by navitoclax, despite the absence of any evidence for a reduction in senescent cell mass in fat. Collectively, this preclinical evidence appears to support the inclusion of senolytics as a component of cancer therapeutics, both to reduce tumor cell mass by the killing of senescent tumor cells, and by collaterally suppressing elements of normal tissue injury derived from the accumulation of non-malignant senescent cells [138]. This can be further encouraged, if senolytics were applied in a temporary fashion (separate pulses following the completion of chemotherapy treatment) in order to cull the burden, instead of the complete eradication, of senescent cells, in a manner that will avoid prolonged use of senolytics. However, in contrast to the hoped-for improvement in the efficacy of chemotherapy coupled with senolytics, the induction of TIS, of itself, was reported to improve outcomes in a cyclophosphamide-treated BCL-2 lymphoma mouse model; it should be noted, though, that evidence for senescence induction was extrapolated indirectly based on the functional status of the INK4a locus in tumor cells [139]. Moreover, senescence induction in colorectal cancer patients in response to 5-fluoruracil was correlated with higher progression-free survival, despite limited sample size [140]; similarly, daunorubicin- and cytarabine-induced senescence in acute myeloid leukemia patients was associated with better disease-free and overall survival states [141]. These in vivo findings coupled with the pre-clinical evidence clearly indicates that we still have an incomplete understanding of the nature of the contribution of senescence to overall cancer treatment outcomes, and whether senescence is ultimately a favorable tumor response to treatment.
2.3. Early Evidence on Senolytics as Anti-Cancer Therapies
By far, the most successful senolytic in preclinical cancer models has been navitoclax. Navitoclax has shown a remarkable capacity to eliminate tumor cells induced into senescence by a variety of therapies, including lung and melanoma cells treated with aurora kinase inhibitors or etoposide [142]; lung cancer cells treated with etoposide or radiation; breast cancer cells treated with doxorubicin, radiation, or BET inhibitors [143][144]; ovarian cancer cells treated with PARP inhibitors; and prostate tumor cells treated with PARP inhibitors or radiation [145]. Clearance of senescent tumor cells by navitoclax has been reported to enhance tumor regression/control and increase mouse survival [143]. As in non-tumor models, the senolytic activity of ABT-263 appears to depend upon inhibition of BCL-XL . Further, our laboratory has recently reported that BAX, but not BAK, is required for cell killing by ABT-263. However, in a recent publication with irradiated soft-tissue sarcoma, BCL-2 inhibition, albeit using supraclinical concentrations of ABT-199, was sufficient to promote senolysis, demonstrating the complex heterogeneity in which BCL-2 family member proteins senescent tumor cells depend upon for survival [146].
Despite the clear effectiveness of navitoclax in pre-clinical studies, navitoclax’s senolytic activity can be highly variable. For example, while prostate cancer LNCaP cells treated with antiandrogens enter senescence, the senescent cells are not eliminated by ABT-263 [147]; however, when treated with PARP inhibitors or radiation, the prostate tumor cells are, in fact, sensitive to senolytic action [145]. The authors have postulated that ABT-263 may only be effective in TIS systems associated with DNA-damage, a provocative concept that will require further studies for confirmation [145].
Another potential drawback to the use of senolytics such as navitoclax is that sensitivity to navitoclax is highly dependent on the expression levels of various BCL-2 family members, which can vary even within cancer cells of the same tumor type undergoing treatment with an identical therapy. This mechanistic element was highlighted by Shahbandi et al., whose work demonstrated that certain breast cancer cell lines remained insensitive to navitoclax following doxorubicin-induced senescence due to low levels of NOXA, a BH3-only protein that can neutralize the anti-apoptotic protein MCL-1 [143]. We have also observed this variability in lung cancer cell line sensitivity to navitoclax upon the promotion of senescence (unpublished data). Finally, the utilization of navitoclax in the clinic may be limited due to the development of severe thrombocytopenia in patients. This toxicity appears to be a consequence of on-target BCL-XL inhibition in platelets [148]. Because navitoclax’s senolytic activity is (often) dependent on BCL-XL, this toxicity appears to be unavoidable without advanced strategies to deliver the drug only to malignant cells. In addition to its known hematologic adverse reactions, navitoclax, when used at effectively senolytic doses, has been shown to cause detrimental bone changes such as decreased trabecular bone volume fraction of mice and impaired osteoblast function in vitro and in vivo, adding another layer of complexity for the use of navitoclax as a senolytic [149]. Its noteworthy that the last discussed evidence is from a single report, where similar adverse effects were not remarkably reported in clinical trials evaluating navitoclax. Lastly, the discussed evidence is largely based on short-term studies of these drugs as senolytics, overlooking any potential toxicities that can arise following chronic use of senolytics.
Although it might be tempting to propose the use of alternative senolytics in the clinic, the cancer field has yet to provide unequivocal pre-clinical support for any senolytic as an adjuvant therapy. There is far too little literature relating to the ability of other compounds, such as D+Q, piperlongumine, and fisetin to act as senolytics in the context of chemotherapy-induced senescence, and what is currently available is not particularly promising. For example, as opposed to its success in the context of aging-related processes, D+Q failed to kill senescent liver cancer cells, and each drug alone failed to kill senescent ovarian cancer cells [150]. Piperlongumine, while showing modest senolytic activity in olaparib-treated ovarian cancer, failed to kill senescent prostate cells. Fisetin failed to kill senescent ovarian cancer cells, and, in our hands, was also largely ineffective against senescent lung, head and neck, and prostate cancer cells (unpublished data). Thus, similar to navitoclax, the senolytic activity of these agents may prove to be highly dependent on the cell lineage and senescence-inducing agents used.
The senolytic activity of a number of other agents that have been identified in tumor models, such as cardiac glycosides [151][152] and the HDAC inhibitor panobinostat [153] must also be considered with reservations. While cardiac glycosides have indeed been shown to eliminate senescent tumor and non-tumor cells, with claims of “broad-spectrum” activity, unfortunately these studies utilized supraphysiological concentrations of these agents that would pose intolerable toxicity to patients [151][152]. Also, there has been only a single report of panobinostat acting as a senolytic in head and neck and lung cancer cell lines induced into senescence by either cisplatin or taxol [153]. Interestingly, and perhaps unexpectedly, panobinostat was also shown, at certain concentrations, to promote senescence when administered in combination with cisplatin or taxol [153].
The HSP90 inhibitor 17-DMAG and the BET protein family degrader ARV825 [154] have both shown broad spectrum activity across senolytic models, including TIS in cancer cells. However, data on these agents is limited, and HSP90 inhibitors can induce intolerable toxicity [155]. BET degraders, while presumably less toxic than BET inhibitors and seemingly “safe” in mice, have yet to be tested in humans[156]. It is possible, however, that the application of these drugs as senolytics may permit the use of lower doses, thereby reducing adverse events. Intriguingly, although ARV825 was proposed to induce senolysis in an autophagy-dependent manner [154], inhibition of autophagy has also been shown to promote senescent cell killing, due to elevated dependence on autophagy caused by proteotoxic stress [157]. Overall, taken together, the currently available evidence is consistent with the lack of universality for any individual senolytic amongst different therapy-induced senescent tumor cell models.
Another potential limitation to the utilization of senolytics is an apparent resistance of some senescent cell subpopulations. For example, while navitoclax has been shown to eliminate approximately 90% of SA-β-gal positive lung and breast tumor cells, there did appear to be residual surviving tumor cells that could reflect a resistant subpopulation . While repetitive exposure to navitoclax (or other senolytics) might prove to eliminate these cells, possible heterogeneity of the senescent phenotype even within a monoclonally derived tumor cell population might confer resistance to senolysis. This analysis is based on data derived from studies in replicative exhaustion-induced senescent fibroblasts, where several distinct transcriptomic profiles can be identified within the same cell line associated with different phenotypic cell behaviors . Post-senolysis, senolytic-resistant senescent cells could contribute to the proliferative recovery of a tumor cell population, over time, following the removal of a senolytic agent . In all cases, the persistence of post-senolytic senescent cells is expected if senolytics are to be utilized in pulses to cull senescent cells, rather than their full eradication.
Finally, it has long been recognized that senescent cells contribute to a number of fundamental homeostatic processes. For example, senescence is a major tumor suppressor mechanism that halts the transformation of cells harboring carcinogenic mutations or oncogene overexpression [158]. So long as senolytics kill senescent cells, rather than hinder the ability to enter the senescent growth arrest, there is little concern that they might be permissive for malignant transformation. Furthermore, although oncogene-induced senescence may be tumor suppressive by one interpretation, accumulation of either oncogene-induced or natural aging related senescence may promote and/or support tumorigenesis through the SASP[159]. This notion is supported by studies from Baker et al., in which aged INK-ATTAC mice that were cleared of p16-positive cells showed increased latency to lethal cancer development [160]. As such, elimination of oncogene- or aging-induced senescent cells are likely of little concern to patient health and would certainly be in-line with senolytic efforts to mitigate aging-related pathological processes. On the other hand, embryonal senescent cells play an important role during organogenesis through paracrine communication with differentiating cells [161]. It is not certain whether these cells persist or carry other physiological roles during adult life. It is also not certain whether they will inadvertently be eliminated by senolytics, especially in that these variants do not show DNA damage or depend on classical cell cycle regulators for their growth arrest. Senescent cells also accumulate at wound sites and participate in the acute inflammatory reaction that mediates wound healing. Moreover, the removal of p16INK4a-positive cells from active wounds using the p16-3MR model interferes with the healing process [162]. This suggests that these cells might also be eliminated in parallel with senescent tumor cells and could result in unwanted impairment of wound healing. Lastly, senescence is involved in recruiting a T cell-mediated immune response against certain viral infections [163], and non-selective removal of these cells by senolytics can potentially increase the risk of viral replication. Overall, the inability of senolytics to differentiate between pathological and physiological senescent cells could represent an additional limitation to their use as cancer therapeutics.
Given the current state of the literature, we have identified four significant challenges (Figure 1) that should be addressed as research continues: (i) the lack of universality of the current senolytics and the need for the identification of the conditions under which each agent is best used, (ii) the selectivity of senolytics towards “harmful”, rather than “beneficial”, senescent cells, (iii) strategies to avoid toxicity associated with the senolytics and (iv) identification of potential resistance mechanisms.
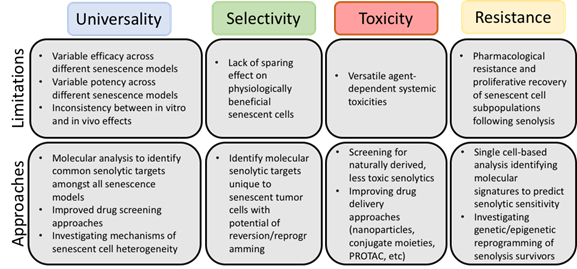
Figure 1. Limitations of current senolytics and potential approaches for circumvention. Although senolytic agents have shown promise as adjuvant cancer therapies preclinically, careful review of the literature has elucidated common limitations.
References
- Karlseder, J.; Smogorzewska, A.; De Lange, T. Senescence Induced by Altered Telomere State, Not Telomere Loss. Sci-ence 2002, 295, 2446–2449, doi:10.1126/science.1069523.
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impair-ment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senes-cence. Autophagy 2017, 13, 99–113, doi:10.1080/15548627.2016.1247143.
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.F.M.; Schlegelberger, B.; Stein, H.; Dörken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nat. Cell Biol. 2005, 436, 660–665, doi:10.1038/nature03841.
- Gewirtz, D.A.; Holt, S.E.; Elmore, L.W. Accelerated senescence: An emerging role in tumor cell response to chemother-apy and radiation. Biochem. Pharmacol. 2008, 76, 947–957, doi:10.1016/j.bcp.2008.06.024.
- Park, J.T.; Lee, Y.-S.; Cho, K.A.; Park, S.C. Adjustment of the lysosomal-mitochondrial axis for control of cellular senes-cence. Ageing Res. Rev. 2018, 47, 176–182, doi:10.1016/j.arr.2018.08.003.
- Chandra, T.; Ewels, P.A.; Schoenfelder, S.; Furlan-Magaril, M.; Wingett, S.W.; Kirschner, K.; Thuret, J.-Y.; Andrew, E.P.; Fraser, P.; Reik, W. Global Reorganization of the Nuclear Landscape in Senescent Cells. Cell Rep. 2015, 10, 471–483, doi:10.1016/j.celrep.2014.12.055.
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated (beta)-galactosidase reflects an increase in lyso-somal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113, 3613–3622.
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senes-cence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 2006, 5, 187–195, doi:10.1111/j.1474-9726.2006.00199.x.
- You, R.; Dai, J.; Zhang, P.; Barding, J.G.A.; Raftery, D. Dynamic Metabolic Response to Adriamycin-Induced Senescence in Breast Cancer Cells. Metabolites 2018, 8, 95, doi:10.3390/metabo8040095.
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial Dysfunction Accounts for the Stochastic Heterogeneity in Telomere-Dependent Senescence. PLoS Bi-ol. 2007, 5, e110, doi:10.1371/journal.pbio.0050110.
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; Zuffo, R.D.; Matti, V.; D’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302, doi:10.1038/ncb2170.
- Wiley, C.D.; Flynn, J.M.; Morrissey, C.; Lebofsky, R.; Shuga, J.; Dong, X.; Unger, M.A.; Vijg, J.; Melov, S.; Campisi, J. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 2017, 16, 1043–1050, doi:10.1111/acel.12632.
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.N.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senes-cence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301, doi:10.1371/journal.pbio.0060301.
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118, doi:10.1146/annurev-pathol-121808-102144.
- Hernandez-Segura, A.; De Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; DeMaria, M. Unmasking Transcriptional Het-erogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4, doi:10.1016/j.cub.2017.07.033.
- Freund, A.; Laberge, R.-M.; DeMaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075, doi:10.1091/mbc.e11-10-0884.
- Barascu, A.; Le Chalony, C.; Pennarun, G.; Genet, D.; Imam, N.; Lopez, B.; Bertrand, P. Oxidative stress induces an ATM-independent senescence pathway through p38 MAPK-mediated lamin B1 accumulation. EMBO J. 2012, 31, 1080–1094, doi:10.1038/emboj.2011.492.
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599, doi:10.1371/journal.pbio.3000599.
- Coppé, J.-P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.-Y.; Campisi, J. Tumor Suppressor and Aging Biomarker p16INK4a Induces Cellular Senescence without the Associated Inflammatory Secretory Phenotype. J. Biol. Chem. 2011, 286, 36396–36403, doi:10.1074/jbc.m111.257071.
- Chen, W.; Wang, X.; Wei, G.; Huang, Y.; Shi, Y.; Li, D.; Qiu, S.; Zhou, B.; Cao, J.; Chen, M.; et al. Single-Cell Transcrip-tome Analysis Reveals Six Subpopulations Reflecting Distinct Cellular Fates in Senescent Mouse Embryonic Fibroblasts. Front. Genet. 2020, 11, 867, doi:10.3389/fgene.2020.00867.
- Tang, H.; Geng, A.; Zhang, T.; Wang, C.; Jiang, Y.; Mao, Z. Single senescent cell sequencing reveals heterogeneity in se-nescent cells induced by telomere erosion. Protein Cell 2018, 10, 370–375, doi:10.1007/s13238-018-0591-y.
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11, S27–S31, doi:10.1016/s0962-8924(01)02151-1.
- Mooi, W.; Peeper, D. Oncogene-Induced Cell Senescence—Halting on the Road to Cancer. N. Engl. J. Med. 2006, 355, 1037–1046, doi:10.1056/nejmra062285.
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Se-nescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822, doi:10.3390/cancers12040822.
- Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P.; Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA Damage Is Able to Induce Senescence in Tumor Cells in Vitro and in Vivo. Cancer Res. 2002, 62, 1876–1883.
- Saleh, T.; Tyutyunyk-Massey, L.; Murray, G.F.; Alotaibi, M.R.; Kawale, A.S.; Elsayed, Z.; Henderson, S.C.; Yakovlev, V.; Elmore, L.W.; Toor, A.; et al. Tumor cell escape from therapy-induced senescence. Biochem. Pharmacol. 2019, 162, 202–212, doi:10.1016/j.bcp.2018.12.013.
- Saleh, T.; Carpenter, V.J.; Tyutyunyk-Massey, L.; Murray, G.; Leverson, J.D.; Souers, A.J.; Alotaibi, M.R.; Faber, A.C.; Reed, J.; Harada, H.; et al. Clearance of therapy-induced senescent tumor cells by the senolytic ABT-263 via interference with BCL-X L –BAX interaction. Mol. Oncol. 2020, 14, 2504–2519, doi:10.1002/1878-0261.12761.
- Li, W.; Wang, W.; Dong, H.; Li, Y.; Li, L.; Han, L.; Han, Z.; Wang, S.; Ma, D.; Wang, H. Cisplatin-induced senescence in ovarian cancer cells is mediated by GRP78. Oncol. Rep. 2014, 31, 2525–2534, doi:10.3892/or.2014.3147.
- Qu, K.; Lin, T.; Wang, Z.; Liu, S.; Chang, H.; Xu, X.; Meng, F.; Zhou, L.; Wei, J.; Tai, M.; et al. Reactive oxygen species generation is essential for cisplatininduced accelerated senescence in hepatocellular carcinoma. Front. Med. 2014, 8, 227–235, doi:10.1007/s11684-014-0327-1.
- Sun, X.; Shi, B.; Zheng, H.; Min, L.; Yang, J.; Li, X.; Liao, X.; Huang, W.; Zhang, M.; Xu, S.; et al. Senescence-associated se-cretory factors induced by cisplatin in melanoma cells promote non-senescent melanoma cell growth through activa-tion of the ERK1/2-RSK1 pathway. Cell Death Dis. 2018, 9, 1–15, doi:10.1038/s41419-018-0303-9.
- Hartman, M.L.; Sztiller-Sikorska, M.; Czyz, M. Whole-exome sequencing reveals novel genetic variants associated with diverse phenotypes of melanoma cells. Mol. Carcinog. 2019, 58, 588–602, doi:10.1002/mc.22953.
- Haferkamp, S.; Borst, A.; Adam, C.; Becker, T.M.; Motschenbacher, S.; Windhövel, S.; Hufnagel, A.L.; Houben, R.; Mei-erjohann, S. Vemurafenib Induces Senescence Features in Melanoma Cells. J. Investig. Dermatol. 2013, 133, 1601–1609, doi:10.1038/jid.2013.6.
- Lee, Y.-H.; Kang, B.S.; Bae, Y.-S. Premature senescence in human breast cancer and colon cancer cells by tamoxi-fen-mediated reactive oxygen species generation. Life Sci. 2014, 97, 116–122, doi:10.1016/j.lfs.2013.12.009.
- Tuttle, R.; Miller, K.R.; Maiorano, J.N.; Termuhlen, P.M.; Gao, Y.; Berberich, S.J. Novel senescence associated gene, YPEL3, is repressed by estrogen in ER+ mammary tumor cells and required for tamoxifen-induced cellular senescence. Int. J. Cancer 2011, 130, 2291–2299, doi:10.1002/ijc.26239.
- Ewald, J.A.; Desotelle, J.A.; Church, D.R.; Yang, B.; Huang, W.; Laurila, T.A.; Jarrard, D.F. Androgen deprivation induces senescence characteristics in prostate cancer cells in vitro and in vivo. Prostate 2012, 73, 337–345, doi:10.1002/pros.22571.
- Blute, M.L.; Damaschke, N.; Wagner, J.; Yang, B.; Gleave, M.; Fazli, L.; Shi, F.; Abel, E.J.; Downs, T.M.; Huang, W.; et al. Persistence of senescent prostate cancer cells following prolonged neoadjuvant androgen deprivation therapy. PLoS ONE 2017, 12, e0172048, doi:10.1371/journal.pone.0172048.
- Barakat, D.J.; Zhang, J.; Barberi, T.; Denmeade, S.R.; Friedman, A.D.; Paz-Priel, I. CCAAT/Enhancer binding protein β controls androgen-deprivation-induced senescence in prostate cancer cells. Oncogene 2015, 34, 5912–5922, doi:10.1038/onc.2015.41.
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treat-ment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122.e12, doi:10.1016/j.ccell.2019.12.006.
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.-Y.J.; Wu, D.Y. Escape from Therapy-Induced Accelerated Cellular Se-nescence in p53-Null Lung Cancer Cells and in Human Lung Cancers. Cancer Res. 2005, 65, 2795–2803, doi:10.1158/0008-5472.can-04-1270.
- Cotarelo, C.L.; Schad, A.; Kirkpatrick, C.J.; Sleeman, J.P.; Springer, E.; Schmidt, M.; Thaler, S. Detection of cellular senes-cence within human invasive breast carcinomas distinguishes different breast tumor subtypes. Oncotarget 2016, 7, 74846–74859, doi:10.18632/oncotarget.12432.
- Chang, B.-D.; Xuan, Y.; Broude, E.V.; Zhu, H.; Schott, B.; Fang, J.; Roninson, I.B. Role of p53 and p21waf1/cip1 in senes-cence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene 1999, 18, 4808–4818, doi:10.1038/sj.onc.1203078.
- Cairney, C.J.; Bilsland, A.E.; Evans, T.J.; Roffey, J.; Bennett, D.C.; Narita, M.; Torrance, C.J.; Keith, W.N. Cancer cell se-nescence: A new frontier in drug development. Drug Discov. Today 2012, 17, 269–276, doi:10.1016/j.drudis.2012.01.019.
- Lee, S.; Lee, A.J.-S. Cellular senescence: A promising strategy for cancer therapy. BMB Rep. 2019, 52, 35–41, doi:10.5483/bmbrep.2019.52.1.294.
- Elmore, L.W.; Rehder, C.W.; Di, X.; McChesney, P.A.; Jackson-Cook, C.K.; Gewirtz, D.A.; Holt, S.E. Adriamycin-induced Senescence in Breast Tumor Cells Involves Functional p53 and Telomere Dysfunction. J. Biol. Chem. 2002, 277, 35509–35515, doi:10.1074/jbc.m205477200.
- Litwiniec, A.; Gackowska, L.; Helmin-Basa, A.; Żuryń, A.; Grzanka, A. Low-dose etoposide-treatment induces en-doreplication and cell death accompanied by cytoskeletal alterations in A549 cells: Does the response involve senes-cence? The possible role of vimentin. Cancer Cell Int. 2013, 13, 9, doi:10.1186/1475-2867-13-9.
- Puig, P.-E.; Guilly, M.-N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; So-lary, E. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043, doi:10.1016/j.cellbi.2008.04.021.
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979, doi:10.1038/ncb1909.
- Alotaibi, M.; Sharma, K.; Saleh, T.; Povirk, L.F.; Hendrickson, E.A.; Gewirtz, D.A. Radiosensitization by PARP Inhibition in DNA Repair Proficient and Deficient Tumor Cells: Proliferative Recovery in Senescent Cells. Radiat. Res. 2016, 185, 229–245, doi:10.1667/rr14202.1.
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E neg-ative cancers. Nat. Commun. 2017, 8, 15916, doi:10.1038/ncomms15916.
- Fleury, H.; Malaquin, N.; Tu, V.; Gilbert, S.; Martinez, A.; Olivier, M.; Sauriol, A.; Communal, L.; Leclerc-Desaulniers, K.; Carmona, E.; et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell re-versible senescence. Nat. Commun. 2019, 10, 1–15, doi:10.1038/s41467-019-10460-1.
- Yang, L.; Fang, J.; Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017, 3, 17049, doi:10.1038/cddiscovery.2017.49.
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Barbosa, I.A.M.; et al. Senescence-associated reprogramming promotes cancer stemness. Nat. Cell Biol. 2018, 553, 96–100, doi:10.1038/nature25167.
- Jonchère, B.; Vétillard, A.; Toutain, B.; Lam, D.; Bernard, A.C.; Henry, C.; Trécesson, S.D.C.; Gamelin, E.; Juin, P.; Guette, C.; et al. Irinotecan treatment and senescence failure promote the emergence of more transformed and invasive cells that depend on anti-apoptotic Mcl-1. Oncotarget 2014, 6, 409–426, doi:10.18632/oncotarget.2774.
- Pluquet, O.; Abbadie, C.; Coqueret, O. Connecting cancer relapse with senescence. Cancer Lett. 2019, 463, 50–58, doi:10.1016/j.canlet.2019.08.004.
- DeMaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cel-lular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2016, 7, 165–176, doi:10.1158/2159-8290.cd-16-0241.
- Duy, C.; Li, M.; Teater, M.; Meydan, C.; Garrett-Bakelman, F.E.; Lee, T.C.; Chin, C.R.; Durmaz, C.; Kawabata, K.C.; Dhi-molea, E.; et al. Chemotherapy induces senescence-like resilient cells capable of initiating AML recurrence. Cancer Dis-cov. 2021, doi:10.1158/2159-8290.cd-20-1375.
- Sagiv, A.; Krizhanovsky, V. Immunosurveillance of senescent cells: The bright side of the senescence program. Biogeron-tology 2013, 14, 617–628, doi:10.1007/s10522-013-9473-0.
- Laberge, R.-M.; Awad, P.; Campisi, J.; Desprez, P.-Y. Epithelial-Mesenchymal Transition Induced by Senescent Fibro-blasts. Cancer Microenviron. 2011, 5, 39–44, doi:10.1007/s12307-011-0069-4.
- Sato, Y.; Kurose, A.; Ogawa, A.; Ogasawara, K.; Traganos, F.; Darzynkiewicz, Z.; Sawai, T. Diversity of DNA damage re-sponse of astrocytes and glioblastoma cell lines with various p53 status to treatment with etoposide and temozolomide. Cancer Biol. Ther. 2009, 8, 452–457, doi:10.4161/cbt.8.5.7740.
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.E.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658, doi:10.1111/acel.12344.
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 1–14, doi:10.1038/s41467-017-00314-z.
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; DeMaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83, doi:10.1038/nm.4010.
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V. Senescence and senotherapeutics: A new field in cancer therapy. Pharmacol. Ther. 2019, 193, 31–49, doi:10.1016/j.pharmthera.2018.08.006.
- Short, S.; Fielder, E.; Miwa, S.; Von Zglinicki, T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 2019, 41, 683–692, doi:10.1016/j.ebiom.2019.01.056.
- Wang, C.; Vegna, S.; Jin, H.; Benedict, B.; Lieftink, C.; Ramirez, C.; De Oliveira, R.L.; Morris, B.; Gadiot, J.; Wang, W.; et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nat. Cell Biol. 2019, 574, 268–272, doi:10.1038/s41586-019-1607-3.
- Sieben, C.J.; Sturmlechner, I.; Van De Sluis, B.; Van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737, doi:10.1016/j.tcb.2018.04.006.
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Dis-ease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046, doi:10.1158/0008-5472.can-18-3437.
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Ca-saclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or athero-sclerotic mice. Aging Cell 2016, 15, 973–977, doi:10.1111/acel.12458.
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; Van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477, doi:10.1126/science.aaf6659.
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532, doi:10.1038/ncomms14532.
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691, doi:10.1038/ncomms15691.
- Nath, K.A.; O’Brien, D.R.; Croatt, A.J.; Grande, J.P.; Ackerman, A.W.; Nath, M.C.; Yamada, S.; Terzic, A.; Tchkonia, T.; Kirkland, J.L.; et al. The murine dialysis fistula model exhibits a senescence phenotype: Pathobiological mechanisms and therapeutic potential. Am. J. Physiol. Physiol. 2018, 315, F1493–F1499, doi:10.1152/ajprenal.00308.2018.
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is as-sociated with cellular senescence in the brain. Aging Cell 2018, 17, e12840, doi:10.1111/acel.12840.
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Sen, J.M.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728, doi:10.1038/s41593-019-0372-9.
- Parikh, P.; Britt, R.D.; Manlove, L.J.; Wicher, S.A.; Roesler, A.; Ravix, J.; Teske, J.; Thompson, M.A.; Sieck, G.C.; Kirkland, J.L.; et al. Hyperoxia-induced Cellular Senescence in Fetal Airway Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2019, 61, 51–60, doi:10.1165/rcmb.2018-0176oc.
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.; Tchkonia, T.; Krüger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab. 2019, 29, 1061–1077.e8, doi:10.1016/j.cmet.2018.12.008.
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; Van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950, doi:10.1111/acel.12950.
- Dai, H.; Chen, R.; Gui, C.; Tao, T.; Ge, Y.; Zhao, X.; Qin, R.; Yao, W.; Gu, S.; Jiang, Y.; et al. Eliminating senescent chon-drogenic progenitor cells enhances chondrogenesis under intermittent hydrostatic pressure for the treatment of OA. Stem Cell Res. Ther. 2020, 11, 1–18, doi:10.1186/s13287-020-01708-5.
- Chandra, A.; Lagnado, A.B.; Farr, J.N.; Monroe, D.G.; Park, S.; Hachfeld, C.; Tchkonia, T.; Kirkland, J.L.; Khosla, S.; Pas-sos, J.F.; et al. Targeted Reduction of Senescent Cell Burden Alleviates Focal Radiotherapy-Related Bone Loss. J. Bone Miner. Res. 2020, 35, 1119–1131, doi:10.1002/jbmr.3978.
- Wang, H.; Wang, Z.; Huang, Y.; Zhou, Y.; Sheng, X.; Jiang, Q.; Wang, Y.; Luo, P.; Luo, M.; Shi, C. Senolytics (DQ) Miti-gates Radiation Ulcers by Removing Senescent Cells. Front. Oncol. 2020, 9, 1576, doi:10.3389/fonc.2019.01576.
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256, doi:10.1038/s41591-018-0092-9.
- Hickson, L.J.; Prata, L.G.L.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456, doi:10.1016/j.ebiom.2019.08.069.
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; Lebrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563, doi:10.1016/j.ebiom.2018.12.052.
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of an-ti-apoptotic factors. Aging Cell 2016, 15, 428–435, doi:10.1111/acel.12445.
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008, 68, 3421–3428, doi:10.1158/0008-5472.can-07-5836.
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190, doi:10.1038/ncomms11190.
- Kim, H.-N.; Chang, J.; Shao, L.; Han, L.; Iyer, S.; Manolagas, S.C.; O’Brien, C.A.; Jilka, R.L.; Zhou, D.; Almeida, M. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell 2017, 16, 693–703, doi:10.1111/acel.12597.
- Pan, J.; Li, D.; Xu, Y.; Zhang, J.; Wang, Y.; Chen, M.; Lin, S.; Huang, L.; Chung, E.J.; Citrin, D.E.; et al. Inhibition of Bcl-2/xl With ABT-263 Selectively Kills Senescent Type II Pneumocytes and Reverses Persistent Pulmonary Fibrosis Induced by Ionizing Radiation in Mice. Int. J. Radiat. Oncol. 2017, 99, 353–361, doi:10.1016/j.ijrobp.2017.02.216.
- Mikawa, R.; Suzuki, Y.; Baskoro, H.; Kanayama, K.; Sugimoto, K.; Sato, T.; Sugimoto, M. Elimination of p19ARF-expressing cells protects against pulmonary emphysema in mice. Aging Cell 2018, 17, e12827, doi:10.1111/acel.12827.
- Xu, X.; Kim, J.J.; Li, Y.; Xie, J.; Shao, C.; Wei, J. Oxidative stress-induced miRNAs modulate AKT signaling and promote cellular senescence in uterine leiomyoma. J. Mol. Med. 2018, 96, 1095–1106, doi:10.1007/s00109-018-1682-1.
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; Van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells pre-vents tau-dependent pathology and cognitive decline. Nat. Cell Biol. 2018, 562, 578–582, doi:10.1038/s41586-018-0543-y.
- Yabluchanskiy, A.; Tarantini, S.; Balasubramanian, P.; Kiss, T.; Csipo, T.; Fülöp, G.A.; Lipecz, A.; Ahire, C.; DelFavero, J.; Nyul-Toth, A.; et al. Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation–induced impairment of neurovascular coupling responses protecting cognitive function in mice. Geroscience 2020, 42, 409–428, doi:10.1007/s11357-020-00154-8.
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, A.; Arthur, H.M.; Passos, J.F.; Richardson, G.D. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 2019, 18, e12945, doi:10.1111/acel.12945.
- Dookun, E.; Walaszczyk, A.; Redgrave, R.; Palmowski, P.; Tual-Chalot, S.; Suwana, A.; Chapman, J.; Jirkovsky, E.; Sosa, L.D.; Gill, E.; et al. Clearance of senescent cells during cardiac ischemia–reperfusion injury improves recovery. Aging Cell 2020, 19, e13249, doi:10.1111/acel.13249.
- Jia, K.; Dai, Y.; Liu, A.; Li, X.; Wu, L.; Lu, L.; Bao, Y.; Jin, Q. Senolytic Agent Navitoclax Inhibits Angiotensin II-Induced Heart Failure in Mice. J. Cardiovasc. Pharmacol. 2020, 76, 452–460, doi:10.1097/fjc.0000000000000878.
- Van Der Feen, D.E.; Bossers, G.P.L.; Hagdorn, Q.A.J.; Moonen, J.-R.; Kurakula, K.; Szulcek, R.; Chappell, J.; Vallania, F.; Donato, M.; Kok, K.; et al. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci. Transl. Med. 2020, 12, 4974, doi:10.1126/scitranslmed.aaw4974.
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; Van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142.e4, doi:10.1016/j.cmet.2019.05.006.
- Sessions, G.A.; Copp, M.E.; Liu, J.; Sinkler, M.A.; D’Costa, S.; Diekman, B.O. Controlled induction and targeted elimina-tion of p16 INK4a -expressing chondrocytes in cartilage explant culture. FASEB J. 2019, 33, 12364–12373, doi:10.1096/fj.201900815rr.
- Yang, H.; Chen, C.; Chen, H.; Duan, X.; Li, J.; Zhou, Y.; Zeng, W.; Yang, L. Navitoclax (ABT263) reduces inflammation and promotes chondrogenic phenotype by clearing senescent osteoarthritic chondrocytes in osteoarthritis. Aging 2020, 12, 12750–12770, doi:10.18632/aging.103177.
- Chung, L.; Maestas, D.R.; Lebid, A.; Mageau, A.; Rosson, G.D.; Wu, X.; Wolf, M.T.; Tam, A.J.; Vanderzee, I.; Wang, X.; et al. Interleukin 17 and senescent cells regulate the foreign body response to synthetic material implants in mice and hu-mans. Sci. Transl. Med. 2020, 12, 3799, doi:10.1126/scitranslmed.aax3799.
- Sugihara, H.; Teramoto, N.; Nakamura, K.; Shiga, T.; Shirakawa, T.; Matsuo, M.; Ogasawara, M.; Nishino, I.; Matsuwaki, T.; Nishihara, M.; et al. Cellular senescence-mediated exacerbation of Duchenne muscular dystrophy. Sci. Rep. 2020, 10, 1–17, doi:10.1038/s41598-020-73315-6.
- Eradat, H.; Grosicki, S.; Catalono, J.; Cosolo, W.; Dyagil, I.; Kipps, T.J.; Zheng, B.; Yalamanchili, S.; Sahasranaman, S.; Hurst, D.; et al. Preliminary Results of a Phase II Open-Label, Randomized Study of the BH3 Mimetic Protein Navitoclax (ABT-263) with or without Rituximab for Treatment of Previously Untreated B-Cell Chronic Lymphocytic Leukemia. Blood 2012, 120, 190, doi:10.1182/blood.v120.21.190.190.
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial Susceptibility of Chronic Lymphocytic Leukemia to BCL2 Inhibition: Results of a Phase I Study of Navitoclax in Patients With Relapsed or Refractory Disease. J. Clin. Oncol. 2012, 30, 488–496, doi:10.1200/jco.2011.34.7898.
- Anuar, N.N.M.; Hisam, N.S.N.; Liew, S.L.; Ugusman, A. Clinical Review: Navitoclax as a Pro-Apoptotic and An-ti-Fibrotic Agent. Front. Pharmacol. 2020, 11, 1817, doi:10.3389/fphar.2020.564108.
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28, doi:10.1016/j.ebiom.2018.09.015.
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963, doi:10.18632/aging.101202.
- Wang, Y.; Chang, J.; Liu, X.; Zhang, X.; Zhang, S.; Zhang, X.; Zhou, D.; Zheng, G. Discovery of piperlongumine as a po-tential novel lead for the development of senolytic agents. Aging 2016, 8, 2915–2926, doi:10.18632/aging.101100.
- Cherif, H.; Bisson, D.G.; Jarzem, P.; Weber, M.; Ouellet, J.; Haglund, L. Curcumin and o-Vanillin Exhibit Evidence of Senolytic Activity in Human IVD Cells In Vitro. J. Clin. Med. 2019, 8, 433, doi:10.3390/jcm8040433.
- Li, W.; He, Y.; Zhang, R.; Zheng, G.; Zhou, D. The curcumin analog EF24 is a novel senolytic agent. Aging 2019, 11, 771–782, doi:10.18632/aging.101787.
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889, doi:10.1038/nrc2961.
- Keren, A.; Tamir, Y.; Bengal, E. The p38 MAPK signaling pathway: A major regulator of skeletal muscle development. Mol. Cell. Endocrinol. 2006, 252, 224–230, doi:10.1016/j.mce.2006.03.017.
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511, doi:10.1038/nrc3057.
- Chakradeo, S.; Elmore, L.W.; Gewirtz, D.A. Is Senescence Reversible? Curr. Drug Targets 2016, 17, 460–466, doi:10.2174/1389450116666150825113500.
- Elmore, L.W.; Di, X.; Dumur, C.; Holt, S.E.; Gewirtz, D.A. Evasion of a Single-Step, Chemotherapy-Induced Senescence in Breast Cancer Cells: Implications for Treatment Response. Clin. Cancer Res. 2005, 11, 2637–2643, doi:10.1158/1078-0432.ccr-04-1462.
- Huang, D.; Leslie, K.A.; Guest, D.; Yeshcheulova, O.; Roy, I.J.; Piva, M.; Moriceau, G.; Zangle, T.A.; Lo, R.S.; Teitell, M.A.; et al. High-Speed Live-Cell Interferometry: A New Method for Quantifying Tumor Drug Resistance and Heterogeneity. Anal. Chem. 2018, 90, 3299–3306, doi:10.1021/acs.analchem.7b04828.
- Guillon, J.; Petit, C.; Moreau, M.; Toutain, B.; Henry, C.; Roché, H.; Bonichon-Lamichhane, N.; Salmon, J.P.; Lemonnier, J.; Campone, M.; et al. Regulation of senescence escape by TSP1 and CD47 following chemotherapy treatment. Cell Death Dis. 2019, 10, 1–19, doi:10.1038/s41419-019-1406-7.
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellu-lar reprogramming in vivo. Science 2016, 354, 4445, doi:10.1126/science.aaf4445.
- Bojko, A.; Czarnecka-Herok, J.; Charzyńska, A.; Dabrowski, M.; Sikora, E. Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells 2019, 8, 1501, doi:10.3390/cells8121501.
- Prata, L.G.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275, doi:10.1016/j.smim.2019.04.003.
- Brighton, P.J.; Maruyama, Y.; Fishwick, K.; Vrljicak, P.; Tewary, S.; Fujihara, R.; Muter, J.; Lucas, E.S.; Yamada, T.; Woods, L.; et al. Clearance of senescent decidual cells by uterine natural killer cells in cycling human endometrium. eLife 2017, 6, e31274, doi:10.7554/elife.31274.
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551, doi:10.1038/nature10599.
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-Cell-Autonomous Tumor Suppression by p53. Cell 2013, 153, 449–460, doi:10.1016/j.cell.2013.03.020.
- Ruscetti, M.; Morris, J.P.; Mezzadra, R.; Russell, J.; Leibold, J.; Romesser, P.B.; Simon, J.; Kulick, A.; Ho, Y.-J.; Fennell, M.; et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 2020, 181, 424–441.e21, doi:10.1016/j.cell.2020.03.008.
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. p53-dependent chemokine production by senes-cent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 2013, 210, 2057–2069, doi:10.1084/jem.20130783.
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senes-cence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660, doi:10.1038/nature05529.
- Kasakovski, D.; Xu, L.; Li, Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. On-col. 2018, 11, 1–9, doi:10.1186/s13045-018-0629-x.
- Malaguarnera, L.; Ferlito, L.; Di Mauro, S.; Imbesi, R.M.; Scalia, G. Immunosenescence and cancer: A review. Arch. Ger-ontol. Geriatr. 2001, 32, 77–93, doi:10.1016/s0167-4943(01)00087-5.
- Ye, J.; Ma, C.; Hsueh, E.C.; Dou, J.; Mo, W.; Liu, S.; Han, B.; Huang, Y.; Zhang, Y.; Varvares, M.A.; et al. TLR 8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol. Med. 2014, 6, 1294–1311, doi:10.15252/emmm.201403918.
- Ramello, M.C.; Tosello, J.; Canale, F.; Mena, H.A.; Negrotto, S.; Gastman, B.; Gruppi, A.; Acosta-Rodriguez, E.; Montes, C.L. Tumor-induced senescent T cells promote the secretion of pro-inflammatory cytokines and angiogenic factors by human monocytes/macrophages through a mechanism that involves Tim-3 and CD40L. Cell Death Dis. 2014, 5, e1507, doi:10.1038/cddis.2014.451.
- Montes, C.L.; Chapoval, A.I.; Nelson, J.; Orhue, V.; Zhang, X.; Schulze, D.H.; Strome, S.E.; Gastman, B.R. Tumor-Induced Senescent T Cells with Suppressor Function: A Potential Form of Tumor Immune Evasion. Cancer Res. 2008, 68, 870–879, doi:10.1158/0008-5472.can-07-2282.
- Ruhland, M.K.; Loza, A.J.; Capietto, A.-H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Com-mun. 2016, 7, 11762, doi:10.1038/ncomms11762.
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing Chemotherapy Efficacy in Pten -Deficient Prostate Tumors by Activating the Senescence-Associated Anti-tumor Immunity. Cell Rep. 2014, 9, 75–89, doi:10.1016/j.celrep.2014.08.044.
- Pereira, B.I.; Devine, O.P.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.J.; Kinsler, V.; Valdovinos, A.; et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8+ T cell in-hibition. Nat. Commun. 2019, 10, 1–13, doi:10.1038/s41467-019-10335-5.
- Muñoz, D.P.; Yannone, S.M.; Daemen, A.; Sun, Y.; Vakar-Lopez, F.; Kawahara, M.; Freund, A.M.; Rodier, F.; Wu, J.D.; Desprez, P.-Y.; et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemothera-py-resistant cancer to aging. JCI Insight 2019, 4, e124716, doi:10.1172/jci.insight.124716.
- Yao, Z.; Murali, B.; Ren, Q.; Luo, X.; Faget, D.V.; Cole, T.; Ricci, B.; Thotala, D.; Monahan, J.; Van Deursen, J.M.; et al. Therapy-Induced Senescence Drives Bone Loss. Cancer Res. 2020, 80, 1171–1182, doi:10.1158/0008-5472.can-19-2348.
- Saleh, T.; Tyutynuk-Massey, L.; Cudjoe, E.K.J.; Idowu, M.O.; Landry, J.W.; Gewirtz, D.A. Non-Cell Autonomous Effects of the Senescence-Associated Secretory Phenotype in Cancer Therapy. Front. Oncol. 2018, 8, 164, doi:10.3389/fonc.2018.00164.
- Acklin, S.; Zhang, M.; Du, W.; Zhao, X.; Plotkin, M.; Chang, J.; Campisi, J.; Zhou, D.; Xia, F. Depletion of senescent-like neuronal cells alleviates cisplatin-induced peripheral neuropathy in mice. Sci. Rep. 2020, 10, 1–11, doi:10.1038/s41598-020-71042-6.
- Sikora, E.; Bielak-Zmijewska, A.; Mosieniak, G. Targeting normal and cancer senescent cells as a strategy of senothera-py. Ageing Res. Rev. 2019, 55, 100941, doi:10.1016/j.arr.2019.100941.
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A Senescence Program Controlled by p53 and p16INK4a Contributes to the Outcome of Cancer Therapy. Cell 2002, 109, 335–346, doi:10.1016/s0092-8674(02)00734-1.
- Haugstetter, A.M.; Loddenkemper, C.; Lenze, D.; Gröne, J.; Standfuss, C.; Petersen, I.; Dörken, B.; Schmitt, C.A. Cellular senescence predicts treatment outcome in metastasised colorectal cancer. Br. J. Cancer 2010, 103, 505–509, doi:10.1038/sj.bjc.6605784.
- Schönlein, M.; Fan, D.N.-Y.; Ihlow, J.; Schwarzer, R.; Vick, B.; Spiekermann, K.; Burmeister, T.; Westermann, J.; Jeremias, I.; Schmitt, C. Therapy-Induced Senescence Is a Predictor of Treatment Outcome in Acute Myeloid Leukemia. Blood 2017, 130, 1393–1393, doi:10.1182/blood.V130.Suppl_1.1393.1393.
- Wang, L.; De Oliveira, R.L.; Wang, C.; Neto, J.M.F.; Mainardi, S.; Evers, B.; Lieftink, C.; Morris, B.; Jochems, F.; Willem-sen, L.; et al. High-Throughput Functional Genetic and Compound Screens Identify Targets for Senescence Induction in Cancer. Cell Rep. 2017, 21, 773–783, doi:10.1016/j.celrep.2017.09.085.
- Shahbandi, A.; Rao, S.G.; Anderson, A.Y.; Frey, W.D.; Olayiwola, J.O.; Ungerleider, N.A.; Jackson, J.G. BH3 mimetics se-lectively eliminate chemotherapy-induced senescent cells and improve response in TP53 wild-type breast cancer. Cell Death Differ. 2020, 27, 3097–3116, doi:10.1038/s41418-020-0564-6.
- Gayle, S.S.; Sahni, J.M.; Webb, B.M.; Weber-Bonk, K.L.; Shively, M.S.; Spina, R.; Bar, E.E.; Summers, M.K.; Keri, R.A. Tar-geting BCL-xL improves the efficacy of bromodomain and extra-terminal protein inhibitors in triple-negative breast cancer by eliciting the death of senescent cells. J. Biol. Chem. 2019, 294, 875–886, doi:10.1074/jbc.ra118.004712.
- Malaquin, N.; Vancayseele, A.; Gilbert, S.; Antenor-Habazac, L.; Olivier, M.; Brahem, Z.A.A.; Saad, F.; Delouya, G.; Rodier, F. DNA Damage- But Not Enzalutamide-Induced Senescence in Prostate Cancer Promotes Senolytic Bcl-xL In-hibitor Sensitivity. Cells 2020, 9, 1593, doi:10.3390/cells9071593.
- Lafontaine, J.; Cardin, G.B.; Malaquin, N.; Boisvert, J.-S.; Rodier, F.; Wong, P. Senolytic Targeting of Bcl-2 Anti-Apoptotic Family Increases Cell Death in Irradiated Sarcoma Cells. Cancers 2021, 13, 386, doi:10.3390/cancers13030386.
- Pungsrinont, T.; Sutter, M.F.; Ertingshausen, M.C.C.M.; Lakshmana, G.; Kokal, M.; Khan, A.S.; Baniahmad, A. Senolytic compounds control a distinct fate of androgen receptor agonist- and antagonist-induced cellular senescent LNCaP pros-tate cancer cells. Cell Biosci. 2020, 10, 1–13, doi:10.1186/s13578-020-00422-2.
- Suvarna, V.; Singh, V.; Murahari, M. Current overview on the clinical update of Bcl-2 anti-apoptotic inhibitors for cancer therapy. Eur. J. Pharmacol. 2019, 862, 172655, doi:10.1016/j.ejphar.2019.172655.
- Sharma, A.K.; Roberts, R.L.; Benson, R.D.J.; Pierce, J.L.; Yu, K.; Hamrick, M.W.; McGee-Lawrence, M.E. The Senolytic Drug Navitoclax (ABT-263) Causes Trabecular Bone Loss and Impaired Osteoprogenitor Function in Aged Mice. Front. Cell Dev. Biol. 2020, 8, 354, doi:10.3389/fcell.2020.00354.
- Kovacovicova, K.; Skolnaja, M.; Heinmaa, M.; Mistrik, M.; Pata, P.; Pata, I.; Bartek, J.; Vinciguerra, M. Senolytic Cocktail Dasatinib+Quercetin (D+Q) Does Not Enhance the Efficacy of Senescence-Inducing Chemotherapy in Liver Cancer. Front. Oncol. 2018, 8, 459, doi:10.3389/fonc.2018.00459.
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 1–12, doi:10.1038/s41467-019-12888-x.
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088, doi:10.1038/s42255-019-0122-z.
- Samaraweera, L.; Adomako, A.; Rodriguez-Gabin, A.; McDaid, H. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 2017, 7, 1900, doi:10.1038/s41598-017-01964-1.
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 2020, 11, 1–13, doi:10.1038/s41467-020-15719-6.
- Neckers, L.; Workman, P. Hsp90 Molecular Chaperone Inhibitors: Are We There Yet? Clin. Cancer Res. 2012, 18, 64–76, doi:10.1158/1078-0432.ccr-11-1000.
- Yang, C.-Y.; Qin, C.; Bai, L.; Wang, S. Small-molecule PROTAC degraders of the Bromodomain and Extra Terminal (BET) proteins—A review. Drug Discov. Today Technol. 2019, 31, 43–51, doi:10.1016/j.ddtec.2019.04.001.
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.M.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425, doi:10.1038/nature12437.
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras Provokes Premature Cell Senescence As-sociated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602, doi:10.1016/s0092-8674(00)81902-9.
- Beck, J.; Turnquist, C.; Horikawa, I.; Harris, C.C. Targeting cellular senescence in cancer and aging: Roles of p53 and its isoforms. Carcinogenesis 2020, 41, 1017–1029, doi:10.1093/carcin/bgaa071.
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.-M.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nat. Cell Biol. 2016, 530, 184–189, doi:10.1038/nature16932.
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Develop-ment. Cell 2013, 155, 1104–1118, doi:10.1016/j.cell.2013.10.019.
- DeMaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733, doi:10.1016/j.devcel.2014.11.012.
- Van Baarle, D.; Tsegaye, A.; Miedema, F.; Akbar, A. Significance of senescence for virus-specific memory T cell respons-es: Rapid ageing during chronic stimulation of the immune system. Immunol. Lett. 2005, 97, 19–29, doi:10.1016/j.imlet.2004.10.003.

