 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sabine Mai | + 3882 word(s) | 3882 | 2021-01-25 06:02:56 |
Video Upload Options
Lamin A/C is a nuclear protein with multiple functions in normal and diseased cells. As lamin A/C has a variety of critical roles within the cell, mutations of the lamin A/C gene and incorrect processing of the protein results in a wide variety of diseases, ranging from striated muscle disorders to accelerated aging diseases.
1. Introduction
Lamin A/C is a protein found in the nuclear lamina of cells[1][2]. It has many roles in cells, from maintaining nuclear shape to regulating gene expression [3][4][5]. As it plays many roles in the cell, it is also involved in various diseases and cancers [6]. One cancer it has been shown to affect is prostate cancer [7][8]. Better understanding of lamin A/C’s role in prostate cancer could help with a more accurate diagnosis of prostate cancer and ensuring patients receive appropriate treatment [7][8].
2. Lamin Proteins
Lamin A/C belongs to the lamin family of proteins [1]. Lamins are 60–80 kDa proteins, which were previously thought to only be found in metazoans, however, lamin-like proteins in non-metazoans, such as NE81 found in Dictyostelium discoideum, have since been discovered [9][10]. Lamins come in two varieties, A-type lamins and B-type lamins [1][11]. Lamin A and lamin C are A-type lamins, commonly referred to together as lamin A/C [1][11]. Lamin A and Lamin C mRNA are produced by the same gene via alternative splicing, and, as they are quite similar, they are frequently studied together and referred to as lamin A/C [12]. While fibroblasts containing only lamin A or lamin C have a slightly abnormal nuclear shape, it appears only either lamin A or lamin C is sufficient for survival as mice made to express only lamin A or lamin C appear normal and healthy [13][14]. At least one A-type lamin is necessary for survival [13][15]. Several mouse models have been made to demonstrate this observation. The mouse model frequently used to show lamin A/C knockdown has exons 8–11 deleted, and these mice typically die 4–8 weeks after birth [15]; however, this model still expresses a truncated form of lamin A [15]. Kim et al. found that LmnaΔ/Δ mice, which have a 357 base pair fragment of the LMNA gene deleted, do not express truncated lamin A/C, and these mice die even sooner, at 16–18 days after birth [15].
Lamin proteins are intermediate filaments [16][17]. Intermediate filaments are divided into six subtypes, of which lamin proteins are type V [1]. Intermediate filaments, which are part of the cellular cytoskeleton, have an average diameter of 10–12 nm, which is in between the diameters of actin and microtubules, hence the name “intermediate” [1][18]. However, at an average diameter of 3.5 nm, a lamin A/C filament is thinner than the average intermediate filament [19]. Intermediate filaments are quite sturdy and can withstand stretching and bending without being damaged; therefore, their role in cells usually has to do with cellular morphology and mechanics [18].
While some lamins are nucleoplasmic, most lamins in the cell are found in the nuclear lamina [12][16][17][20]. The nuclear lamina is a meshwork of A- and B-type lamins found under the inner nuclear membrane [16]. Both A- and B-type nuclear lamin proteins have many different roles in the cell. The nuclear lamina provides structural support to the nucleus, thereby maintaining proper nuclear morphology [11]. The nuclear lamina also links the cytoskeleton to the nucleoskeleton via Linker of Nucleoskeleton and Cytoskeleton (LINC) complexes, which are composed of Sad1/UNC-84 (SUN) domain proteins and Klarischt/ANC-1/Syne homology (KASH) domain proteins [11][21]. The LINC complex SUN1 and SUN 2 proteins can interact with nesprin family proteins found on the outer nuclear membrane, which interacts with the cytoplasm [1]. A- and B-type lamin proteins have many binding partners and form many interactions with various inner nuclear membrane proteins and heterochromatin and accordingly play many different roles in the cell, from chromatin organization to DNA repair [1][16][17][20]. Additionally, it has been found that depending on the tissue type, lamins have different binding partners, which could cause it to play different roles depending on the tissue type [1].
3. Lamin A/C: Gene, Protein, and Nuclear Lamina Structure
The human genome has three genes, which code for lamin proteins; LMNA, LMNB1, and LMNB2 [1][17][20][22]. B-type lamins are encoded by the LMNB1 and LMNB2 genes. A-type lamins are encoded by the gene LMNA, which, along with the major isoforms A and C, also encodes minor isoforms C2 and AΔ10 [1]. Lin and Worman used sequencing and restriction mapping to determine structural organization of the human LMNA gene and found the gene has 12 exons, and the coding region is around 24 kb [12]. Additionally, it was found that lamin A and lamin C are identical up to the 566th amino acid [12]. Alternative splicing occurs at exon 10 to result in mRNA for prelamin A and lamin C [12]. B-type lamins have three subtypes; B1, encoded by LMNB1, and B2 and B3, which are encoded by LMNB2, with B3 being only found in amphibians and fish [1][22].
Lamins have the typical tripartite structure found in other intermediate filaments, made up of a central α helical domain with a N-terminal head and C-terminal tail on either side [1][16][17][20]. The central α helical rod domain is made up of four segments 1A, 1B, 2A, and 2B, connected by subdomains L1, L12, and L2 [1][22]. Unlike other cytoplasmic intermediate filaments, lamins also have a nuclear localization signal, and Ig fold in their tail domain [1][17][20]. The Ig domain is made up of two β sheets and plays a role in protein-protein and protein-ligand interaction [1]. Additionally, lamin A has two extra exons that are spliced out in lamin C, which encode a CAAX box (C = cysteine, A = aliphatic residue, X = any other residue) [1][12][23]. The CAAX box can be modified via farnesylation and allows lamin A’s immature form, prelamin A, to undergo post-translational modifications [1][12][16][23]. B-type lamins also contain a CAAX box, and unlike lamin A, remain permanently farnesylated [22]. As lamin C lacks a CAAX box, it is translated as its mature form and requires no further processing [23].
As LMNA translation and splicing produces prelamin A, which cannot incorporate into the nuclear lamina, it must be post-translationally modified to make mature lamin A [12]. To form lamin A, the cysteine of the CAAX box is first farnesylated by farnesyltransferase, then the last three amino acids of the protein are cleaved by RAS-converting enzyme 1 (RCE1) or Zmpste24, and the farnesylated cysteine is methylated by isoprenylcysteine carboxyl methyltransferase (ICMT) [16][17][23]. It is thought that farnesylation helps B-type lamins and lamin A localize to the nuclear envelope [1]. This is further shown by the fact that during mitosis, B-type lamins, which are permanently farnesylated, remain at the nuclear periphery, while lamin A, which has its farnesyl moiety cleaved, is far more soluble [22]. As lamin C has no CAAX box and, therefore, cannot be farnesylated, lamin C is able to rely on other methods localize to the nuclear envelope [1]. Besides farnesylation, lamin proteins can be targeted to the nuclear envelope via specific membrane receptors, which bind lamins [24]. Additionally, phosphorylation plays a role in lamin A/C incorporation into the nuclear lamina, as well as disassembly during mitosis [25]. It is also thought that lamin A can aid lamin C in nuclear envelope localization [24]. Once the lamin A is localized to the nuclear envelope, Zmpste24 cleaves the last 15 amino acids of the protein, including the farnesylated cysteine, to form mature lamin A [16][17][22][23]. Proper prelamin A processing is important as incorrect processing can result in various diseases [14]. For example, if the final 15 amino acids fail to be cleaved, it can lead to progerin, the permanently farnesylated mutated lamin A found in progeria, an aging disease [17]. Additionally, mutations resulting in a lack of Zmpste24 can also cause restrictive dermopathy, which results in bone and skin defects, among other complications, leading to early death in humans [13]. While Zmpste24−/− mice do not die soon after birth like humans, they do develop muscle weakness, reduced bone density, and slowed growth [13].
While generally lamin A and lamin C levels are equal in cells, differences have been found in some tissues, with neuronal cells appearing to express higher lamin C levels and only lamin C being found in the human skin’s basal layer [5][23]. In mice, neuron and glia cells show a lack of lamin A, and it has been found that the brain-specific microRNA miR-9 only downregulates expression of lamin A but not lamin C in these cell types [14]. Additionally, the lamin A/lamin C ratio has also been shown to be altered in some diseases such as HIV, type 2 diabetes, progeria, and dilated cardiomyopathy [23]. Lamin C is also only found in mammals, while lamin A is found in all vertebrates [23]. Studies have also found that lamin A and lamin C have different binding affinities and partners, one example being SUN1, a nuclear envelope protein, which only binds lamin A [23].
As shown in Figure 1a, lamin A/C is commonly found around the nucleus as part of the nuclear lamina, where it polymerizes to form filaments around the nucleus [1][2]. Some lamin A/C is also found in the nucleoplasm [1]. Nucleoplasmic lamin A/C is not polymerized and is more mobile [1][2][26]. While some are probably bound to structures within the nucleus, the mobile portion of nucleoplasmic lamin A is around 60% [2]. Lamin-associated polypeptide 2α (LAP2α) is a lamin binding partner, which has been found to help in localizing lamin A to the nuclear interior rather than the lamina [2].
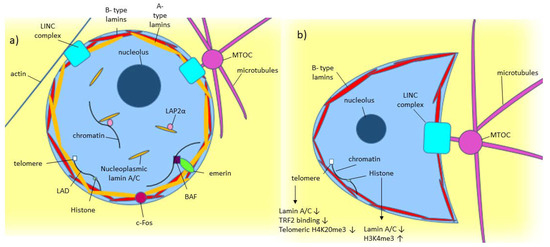
Figure 1. Model of known Lamin A/C functions in normal and diseased cells. (a) A-type lamin’s location and binding partners within the nucleus in normal cells. (b) Effect of low/absent lamin A/C on a cell, including crescent-shaped nuclear morphology caused by the microtubule organizing center (MTOC) pushing into the nucleus [27].
Cryo-electron tomography (Cryo-ET) has shown that the nuclear lamina is around 14 nm thick, and the A- and B- type lamins are found in lamin filaments with alternating tetrameric and hexameric regions [19][28]. To form these tetrameric filaments, lamin proteins first form dimers via coiled-coil interactions of their rod domains [11][22]. In vitro, it has been found that different types of lamins can form heterotypic dimers, but in vivo, it has been found that lamins likely only form homotypic dimers [1][22]. These dimers then assemble end to end, head to tail, to form protofilaments in which the Ig-domains are spaced about 40 nm apart [11][19]. Finally, these protofilaments join laterally to form the final tetrameric filaments found in the nuclear lamina [11].
While in vitro, lamin filaments form paracrystalline arrays, in vivo, it has been shown with confocal microscopy of HeLa cells and 3D-SIM imagining of mouse embryonic fibroblasts that A- and B-type lamins instead form a meshwork [11][19][26]. In mammalian somatic cells, A- and B-type isoforms form separate meshworks in the nuclear lamina with the A-type lamin meshwork facing the nucleoplasm and the B-type lamin meshwork outside of the A-type mesh [5][16][19][26][28]. The meshworks are thought to be separate, but the silencing of one isoform has been shown to affect the meshworks of the remaining isoforms. For example, silencing of lamin B1 led to the enlarging of the lamin A/C mesh [11][26]. Lamin B1 silencing also caused nuclear blebs, found to contain only lamin A/C and no B-type lamins [11][22][26]. It was suggested that this phenomenon occurred because lamin B1 is found outside the lamin A/C meshwork, holding it in, and, therefore, its loss causes the lamin A/C meshwork to expand, causing blebs [5]. As silencing one type of lamin was shown to affect the remaining lamins in the nuclear lamina, it is likely that the different lamin isoforms still interact with each other despite forming separate meshworks [19][26][28]. When it comes to how lamin A is incorporated into the nuclear lamina, one study found that Sorting Nexin 6, which regulates the trafficking of various proteins, plays a role in lamin A import to the nucleus using a RAN-GTP (Ras-related nuclear protein) gradient [29].
During mitosis, the nuclear lamina must disassemble. This is achieved through phosphorylation of the lamina [26]. CDK 1 phosphorylates the lamins during mitosis, and it has been found that A-type lamins leave the nuclear lamina first and are freely diffusible in the nuclear interior, unlike B-type lamins, which remain near the nuclear periphery even after phosphorylation [26]. Protein phosphatase 1 dephosphorylates the lamins when reassembly of the lamina is needed [26]. In addition to phosphorylation, lamins can also undergo various other modifications such as sumoylation, acetylation, and ubiquitylation [1][2]. Lamin A/C acetylation has been found to be important for lamin A/C to function correctly. MOF, a lysine acetyltransferase, which is part of the non-specific lethal (NSL) protein complex acetylates lamin A/C [30]. In MOF knockout mouse MEFs, loss of lamin A/C acetylation results in lamin A/C having increased solubility, which greatly affects its functioning in mechanotransduction and resulted in nuclear blebbing, nuclear envelope rupture, transcriptional defects, and general genomic instability [30].
4. Lamin A/C in Disease and Cancer
4.1. Laminopathies
As lamin A/C has so many different functions, mutations impairing these functions can lead to a wide variety of diseases. Diseases involving lamin proteins are called laminopathies and can be primary or secondary [6][13][31] Primary laminopathies are caused by mutation of the LMNA gene, and secondary laminopathies are caused by Zmpste24 defects, which prevents prelamin A from successfully being converted to lamin A [6][13][31]. Secondary laminopathies can also result from mutation of other non-lamin genes that interact with lamin such as lamin binding proteins like emerin [6][13][31]. Additionally, while it is lamin A/C mutations that most commonly cause laminopathies, lamin B can also be involved, with Pelger-Huet anomaly and Greenberg skeletal dysplasia being caused by a mutation to the LBR gene [22]. Laminopathies can be caused by lamin A/C mutations affecting both the cell’s structural integrity and genomic stability [32][33]. Generally primary laminopathies can be sorted into 4 types; striated muscle disorders, lipodystrophic syndromes, peripheral neuropathy, and accelerated aging diseases [6][22]. Secondary laminopathies have caused mandibuloacral disease, progeroid-like disease, and restrictive dermopathy [13].
The first disorder lamin A/C was ever found to be implicated in was Emery-Dreifuss muscular dystrophy (EDMD) [6]. EDMD primarily affects skeletal muscle, but cardiac muscle can also be affected [6]. LMNA mutations can also lead to dilated cardiomyopathy (DCM). DCM is the third leading cause of heart failure in the United States, and about 6% of cases are caused by LMNA mutations [6][32]. DCM caused by the inheritance of LMNA mutations is usually autosomal dominant and typically associated with a more severe phenotype and poorer prognosis than DCM caused by other causes [34]
Lipodystrophic syndromes cause loss of adipose tissue and insulin resistance [31][35]. As well as lipodystrophic syndromes, lamin A/C is also implicated in obesity-induced insulin resistance [36]. Lamin A/C increases pro-inflammatory NF-κβ transcription factor transcription, which mediates adipose tissue macrophage inflammation and can lead to insulin resistance and type 2 diabetes [36].
LMNA mutations can also cause accelerated aging disorders like Hutchinson-Gilford progeria syndrome (HGPS) and Werner syndrome [31]. 90% of all HGPS is caused by a single base-pair substitution at exon 11 of lamin A, which results in the permanently farnesylated protein progerin [13][31]. Typically the brain is not affected in HGPS patients, but it has been found that LMNA expression in the hippocampus increases in late-stage Alzheimer’s [37]. It is also thought lamin A/C may play a role in regular aging as many of the nuclear defects found in HGPS cells are the same as ones found in cells from older people [37][38].
Currently, laminopathies lack specific treatment. Lipodystrophic syndromes caused by LMNA receive the same treatment as diabetes Type II or dyslipidemia, such as diet, exercise, metformin, and insulin [35]. Additionally, recombination leptin therapy can also relieve some of the symptoms of lipodystrophy, like insulin resistance [36]. As for DCM, treatments are the same as non-LMNA caused heart failure and usually involve pacemakers and heart transplants once the disease becomes severe enough [6]. One potential target for laminopathy-associated cardiomyopathy treatment is LSD1, a H3K4me1 demethylase.[39] A p.H222P Lmna mutation results in autosomal dominant EDMD.[39] Using mouse embryonic stem cells and mice with this mutation Guenantin et al. found that a loss of H3K4me1 on the enhancers of genes involved in epithelial-to-mesenchymal transition and cardiogenesis resulted in defective cardiac development.[39] Inhibiting LSD1 improved cardiogenesis and cardiac function in these cells.[39] Therefore LSD1 inhibitors like GSK-LSD1 could potentially be tested for use in treating laminopathy-associated cardiomyopathy.[39] For HSPG, there have been several studies looking into various potential treatments. One such potential treatment is using farnesyl-transferase inhibitors such as lonafarnib to alleviate problems in HGPS cells, but while lonafarnib has been shown to improve nuclear shape in HGPS cells, it is too toxic for long term treatment [6][40][31]. Another potential HGPS treatment is progerinin (SLC-D011), a modified form of progerin-lamin A binding inhibitor JH4, which is not severely toxic at high concentrations.[41] When tested in mouse models of progeria, progerinin increased mouse body weight, decreased progerin levels, and increased life span for a longer interval than Ionafarnib.[41] In vivo base editing is also being looked into as a potential HGPS treatment.[42] Since HGPS is often caused by a CG to TA mutation in LMNA, adenine base editors (ABE) have been used to try and correct the mutation in fibroblast cell lines derived from progeria patients and progeria mouse models.[42] In the cell lines, ABE reduced progerin levels and improved nuclear morphology.[42] Additionally, cardiovascular disease is a frequent cause of death in progeria patients, and in mouse models, ABE treatment resulted in preserved vascular smooth muscle cell (VSMC) counts and increased life span when compared to saline-injected controls.[42] Another study found that inhibiting isoprenylcysteine carboxymethyltransferase (ICMT), which methylates progerin, could be a potential HGPS treatment.[43] In HGPS mouse models LmnaG609G/G609G and LmnaG609G/+, the knockout of the ICMT gene resulted in longer life span and normalized VSMC numbers.[43] C75, a ICMT inhibitor was also tested on human HGPS cell lines and was found to delay senescence and increase cell proliferation.[43] However, as C75 has poor predicted bioavailability, a new compound would be needed to test ICMT inhibition in vivo.[43] Overall, most current treatments for laminopathies focus on treating symptoms rather than the root cause.
4.2. Cancer
Cancer cells have dysregulated gene expression, alterations in signaling pathways, overall genomic instability, and abnormal nuclear shape [44][45]. As lamin A/C plays many roles in the cell, including regulating gene expression, participating in signaling pathways, and maintaining proper nuclear shape, it is likely lamin A/C also plays a role in the development and/or maintenance and propagation of cancer cells. Cancer cells frequently have irregular lamin A/C expression and, as seen in Table 1, Table 2 and Table 3, lamin A/C can be over or under-expressed in many different cancer types. Additionally, lamin A/C also shows incorrect cytoplasmic localization in some cancer cells as well as abnormal internal lamin A/C structures within the nucleus [44][46][47][48][49]. However, since lamin A/C plays so many different roles in cells and its function can vary across different tissue types, its effect in cancer, as shown by Table 1, Table 2 and Table 3, is highly variable across different cancer subtypes, and there is no overall expression pattern for lamin A/C in cancer [44][48][50]. Additionally, while lamin A/C expression levels are often measured together, some studies have found lamin A and lamin C expression is not always affected equally in some cancers, further complicating lamin A/C’s effect on cancer [51][52][53].
Table 1. Lamin A/C expression effect on different cancer cell lines.
|
Cell and Cancer Type |
Lamin A/C Expression |
Effect on Cancer |
Reference |
|
Lung cancer cell lines A549, and H1299 |
Decrease in lamin A/C |
Increased tumor senescence |
[54] |
|
Human non-small cell lung carcinoma H 1299, human osteosarcoma U-2 OS, human cervix carcinoma HeLa |
Lamin A/C mutated to S22A-progerin |
Increased cellular senescence |
[55] |
|
Breast adenocarcinoma cell lines MDA-MB-231 and MDA-MB-468 |
Lamin A/C knockout |
Decreased cell’s resistance to fluid shear stress |
[56] |
|
Human pre-metastatic colon adenocarcinoma cell line SW480 |
Transfected with GFP-lamin A |
Downregulated E-cadherin and increased cell motility |
[57] |
|
Human pre-metastatic colon adenocarcinoma cell line SW480 |
Lamin A/C upregulated |
Increased cell motility |
[46] |
|
Human ovarian cancer cell line H08910 |
Lamin A/C overexpressed, Lamin A/C 30–40% inhibited, lamin A/C 70–80% inhibited |
Lamin A/C overexpressed: Decreased migration through 3 μm pore. 30–40% inhibited: Increased migration through 3 μm pore. 70–80% inhibited: Decreased migration through 3 μm pore |
[58] |
|
Prostate cancer cell line DU145 and breast cancer cell line BT-549 |
Lamin A/C depleted |
Emerin mislocalized and nuclear membrane had blebbing |
[59] |
|
Neuroblastoma cell line SH-SY5Y |
Lamin A/C downregulated |
Tumor initiating cells developed |
[60] |
|
Breast cancer cell line MDA-MB-231 in suspension culture |
Lamin A/C downregulated |
Decreased adhesion and reattachment of cells |
[61] |
|
Prostate cancer cell line PC3, DU145, and LNCaP |
Lamin A/C upregulated |
Increased PI3k subunit, p110, and p85 expression and increased growth and invasive capabilities |
[62] |
|
Prostate cancer cell line PC-3M-1E8 and PC-3M-2B4 |
Lamin A/C knockdown |
Cell growth was inhibited and colony formation was decreased. E-cadherin downregulated. Vimentin, snail, and slug upregulated |
[63] |
Table 2. Lamin A/C expression found in patient samples.
|
Cancer Type |
Lamin A/C Expression |
Reference |
|
56 invasive ductal carcinoma samples |
Majority of samples heterogeneous for lamin A/C expression, 38% had no lamin A/C expressed |
[64] |
|
656 colorectal adenocarcinoma tumor samples |
70% positive for lamin A/C, 30% negative |
[57] |
|
73 breast cancer tumor samples |
84.9% had low lamin A/C expression (low defined as less than 50% of cancer cells stained positive for lamin A/C) |
[65] |
|
33 small cell lung carcinomas (SCLC), 72 non-small cell lung carcinomas (34 adenocarcinoma, 30 squamous cell carcinoma, 8 large cell carcinoma) |
91% of SCLC had negative or low lamin A/C expression, 3% of non-SCLC had negative or low lamin A/C expression |
[66] |
|
115 breast cancer tissue samples |
Lower lamin A/C than found in non-cancerous breast tissue. |
[67] |
|
126 gastric carcinoma samples |
70 positive for lamin A/C 56 negative for lamin A/C (lamin A/C associated with poorer prognosis and lower differentiation) |
[68] |
|
87 endometrial cancer samples |
Lamin A was reduced in all high-grade endometrial cancer samples |
[69] |
|
61 epithelial ovarian cancer samples |
Lower lamin A expression than normal and benign controls |
[51] |
|
17 primary colorectal carcinomas, 18 adenomatous polyps |
Lamin A/C reduced or absent in all samples |
[47] |
|
128 breast adenocarcinomas |
Lamin C expression increased, Lamin A expression decreased |
[52] |
|
76 cervical uterine smears (CUS) |
39% normal expression, 28% weak, 33% none. Oncogenic HPV infection rate highest in group with no lamin A/C staining. |
[70] |
|
219 stage II and 151 stage III colon cancer samples |
17.8% low lamin A/C expression, Reoccurrence of cancer 45.5% in low lamin A/C expression group compared to 29.6% in high lamin A/C expression group |
[71] |
|
94 prostate tumor tissue microarrays |
Lamin A/C had low expression in tumor regions with a Gleason pattern (GP) less than 3 and higher expression in regions with a GP of 4 or 5 |
[62] |
|
4 prostate adenocarcinoma cohorts |
Lamin A/C mRNA reduced when the Gleason score is 8, but the level increases above Gleason score 8 and in metastatic regions |
[63] |
|
Tissue microarray from 501 prostate cancer patients |
Low lamin A/C associated with increased lymph node metastasis and disease-specific death |
[7] |
|
Tissue cores from 94 prostate tumor samples |
Lamin A expression higher in higher Gleason score tumors |
[72] |
|
Biopsies from 9 patients with Hodgkin’s disease |
Most Reed-Sternberg and Hodgkin cells expressed lamin A/C while surrounding B and T lymphocytes did not. |
[73] |
Table 3. Lamin A/C expression effect on different cancers in patient samples.
|
Cancer Type |
Lamin A/C Expression |
Effect on Cancer |
Reference |
|
Invasive breast carcinoma |
Reduced lamin A/C expression |
More aggressive phenotype than tumors with high lamin A/C expression |
[74] |
|
Stage II and Stage III colon cancer tumors |
Low lamin A/C expression |
Increased disease recurrence |
[71] |
|
Colorectal adenocarcinoma tumors |
Sample either did or did not express lamin A/C |
Mortality risk twice as high as tumors not expressing lamin A/C |
[57] |
References
- Dittmer, T.A.; Misteli, T. The lamin protein family. Genome Biol. 2011, 12, 222.
- Naetar, N.; Ferraioli, S.; Foisner, R. Lamins in the nuclear interior-life outside the lamina. J. Cell Sci. 2017, 130, 2087–2096.
- Lund, E.; Oldenburg, A.R.; Delbarre, E.; Freberg, C.T.; Duband-Goulet, I.; Eskeland, R.; Buendia, B.; Collas, P. Lamin A/C-promoter interactions specify chromatin state-dependent transcription outcomes. Genome Res. 2013, 23, 1580–1589.
- Osmanagic-Myers, S.; Dechat, T.; Foisner, R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015, 29, 225–237.
- Nmezi, B.; Xu, J.; Fu, R.; Armiger, T.J.; Rodriguez-Bey, G.; Powell, J.S.; Ma, H.; Sullivan, M.; Tu, Y.; Chen, N.Y.; et al. Concentric organization of A- and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. Proc. Natl. Acad. Sci. USA 2019, 116, 4307–4315.
- Wang, X.; Zabell, A.; Koh, W.; Tang, W.H. Lamin A/C Cardiomyopathies: Current Understanding and Novel Treatment Strategies. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 21.
- Saarinen, I.; Mirtti, T.; Seikkula, H.; Boström, P.J.; Taimen, P. Differential Predictive Roles of A- and B-Type Nuclear Lamins in Prostate Cancer Progression. PLoS ONE 2015, 10, e0140671.
- Meaburn, K.J.; Misteli, T. Assessment of the Utility of Gene Positioning Biomarkers in the Stratification of Prostate Cancers. Front. Genet. 2019, 10, 1029.
- Preisner, H.; Habicht, J.; Garg, S.G.; Gould, S.B. Intermediate filament protein evolution and protists. Cytoskeleton 2018, 75, 231–243.
- Krüger, A.; Batsios, P.; Baumann, O.; Luckert, E.; Schwarz, H.; Stick, R.; Meyer, I.; Gräf, R. Characterization of NE81, the first lamin-like nucleoskeleton protein in a unicellular organism. Mol. Biol. Cell 2012, 23, 360–370.
- Shimi, T.; Kittisopikul, M.; Tran, J.; Goldman, A.E.; Adam, S.A.; Zheng, Y.; Jaqaman, K.; Goldman, R.D. Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol. Biol. Cell 2015, 26, 4075–4086.
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326.
- Stewart, C.L.; Kozlov, S.; Fong, L.G.; Young, S.G. Mouse models of the laminopathies. Exp. Cell Res. 2007, 313, 2144–2156.
- Jung, H.J.; Lee, J.M.; Yang, S.H.; Young, S.G.; Fong, L.G. Nuclear lamins in the brain-new insights into function and regulation. Mol. Neurobiol. 2013, 47, 290–301.
- Kim, Y.; Zheng, Y. Generation and characterization of a conditional deletion allele for Lmna in mice. Biochem. Biophys. Res. Commun. 2013, 440, 8–13.
- Zwerger, M.; Medalia, O. From lamins to lamina: A structural perspective. Histochem. Cell Biol. 2013, 140, 3–12.
- de Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear Lamins: Thin Filaments with Major Functions. Trends Cell Biol. 2018, 28, 34–45.
- Goldmann, W.H. Intermediate filaments and cellular mechanics. Cell Biol. Int. 2018, 42, 132–138.
- Turgay, Y.; Eibauer, M.; Goldman, A.E.; Shimi, T.; Khayat, M.; Ben-Harush, K.; Dubrovsky-Gaupp, A.; Sapra, K.T.; Goldman, R.D.; Medalia, O. The molecular architecture of lamins in somatic cells. Nature 2017, 543, 261–264.
- Gruenbaum, Y.; Medalia, O. Lamins: The structure and protein complexes. Curr. Opin. Cell Biol. 2015, 32, 7–12.
- Zhang, B.; Yang, Y.; Keyimu, R.; Hao, J.; Zhao, Z.; Ye, R. The role of lamin A/C in mesenchymal stem cell differentiation. J. Physiol. Biochem. 2019, 75, 11–18.
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 2015, 84, 131–164.
- Al-Saaidi, R.; Bross, P. Do lamin A and lamin C have unique roles? Chromosoma 2015, 124, 1–12.
- Pugh, G.E.; Coates, P.J.; Lane, E.B.; Raymond, Y.; Quinlan, R.A. Distinct nuclear assembly pathways for lamins A and C lead to their increase during quiescence in Swiss 3T3 cells. J. Cell Sci. 1997, 110, 2483–2493.
- Torvaldson, E.; Kochin, V.; Eriksson, J.E. Phosphorylation of lamins determine their structural properties and signaling functions. Nucleus 2015, 6, 166–171.
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Goldman, R.D. Nuclear lamins in cell regulation and disease. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 525–531.
- Tariq, Z.; Zhang, H.; Chia-Liu, A.; Shen, Y.; Gete, Y.; Xiong, Z.M.; Tocheny, C.; Campanello, L.; Wu, D.; Losert, W.; et al. Lamin A and microtubules collaborate to maintain nuclear morphology. Nucleus 2017, 8, 433–446.
- Kittisopikul, M.; Virtanen, L.; Taimen, P.; Goldman, R.D. Quantitative Analysis of Nuclear Lamins Imaged by Super-Resolution Light Microscopy. Cells 2019, 8, 361.
- González-Granado, J.M.; Navarro-Puche, A.; Molina-Sanchez, P.; Blanco-Berrocal, M.; Viana, R.; Font de Mora, J.; Andrés, V. Sorting nexin 6 enhances lamin a synthesis and incorporation into the nuclear envelope. PLoS ONE 2014, 9, e115571.
- Karoutas, A.; Szymanski, W.; Rausch, T.; Guhathakurta, S.; Rog-Zielinska, E.A.; Peyronnet, R.; Seyfferth, J.; Chen, H.R.; de Leeuw, R.; Herquel, B.; et al. The NSL complex maintains nuclear architecture stability via lamin A/C acetylation. Nat. Cell Biol. 2019, 21, 1248–1260.
- Kang, S.M.; Yoon, M.H.; Park, B.J. Laminopathies; Mutations on single gene and various human genetic diseases. BMB Rep. 2018, 51, 327–337.
- Tesson, F.; Saj, M.; Uvaize, M.M.; Nicolas, H.; Płoski, R.; Bilińska, Z. Lamin A/C mutations in dilated cardiomyopathy. Cardiol. J. 2014, 21, 331–342.
- Gerbino, A.; Procino, G.; Svelto, M.; Carmosino, M. Role of Lamin A/C Gene Mutations in the Signaling Defects Leading to Cardiomyopathies. Front. Physiol. 2018, 9, 1356.
- Hasselberg, N.E.; Haland, T.F.; Saberniak, J.; Brekke, P.H.; Berge, K.E.; Leren, T.P.; Edvardsen, T.; Haugaa, K.H. Lamin A/C cardiomyopathy: Young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 2018, 39, 853–860.
- Vigouroux, C.; Guénantin, A.C.; Vatier, C.; Capel, E.; Le Dour, C.; Afonso, P.; Bidault, G.; Béréziat, V.; Lascols, O.; Capeau, J.; et al. Lipodystrophic syndromes due to LMNA mutations: Recent developments on biomolecular aspects, pathophysiological hypotheses and therapeutic perspectives. Nucleus 2018, 9, 235–248.
- Kim, Y.; Bayona, P.W.; Kim, M.; Chang, J.; Hong, S.; Park, Y.; Budiman, A.; Kim, Y.J.; Choi, C.Y.; Kim, W.S.; et al. Macrophage Lamin A/C Regulates Inflammation and the Development of Obesity-Induced Insulin Resistance. Front. Immunol. 2018, 9, 696.
- Méndez-López, I.; Blanco-Luquin, I.; Sánchez-Ruiz de Gordoa, J.; Urdánoz-Casado, A.; Roldán, M.; Acha, B.; Echavarri, C.; Zelaya, V.; Jericó, I.; Mendioroz, M. Hippocampal LMNA Gene Expression is Increased in Late-Stage Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 878.
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063.
- Anne-Claire Guénantin; Imen Jebeniani; Julia Leschik; Erwan Watrin; Gisèle Bonne; Nicolas Vignier; Michel Pucéat; Targeting the histone demethylase LSD1 prevents cardiomyopathy in a mouse model of laminopathy. Journal of Clinical Investigation 2021, 131, e136488, 10.1172/jci136488.
- Gonzalo, S.; Kreienkamp, R.; Askjaer, P. Hutchinson-Gilford Progeria Syndrome: A premature aging disease caused by LMNA gene mutations. Ageing Res. Rev. 2017, 33, 18–29.
- So-Mi Kang; Min-Ho Yoon; Jinsook Ahn; Ji-Eun Kim; So Young Kim; Seock Yong Kang; Jeongmin Joo; Soyoung Park; Jung-Hyun Cho; Tae-Gyun Woo; et al.Ah-Young OhKyu Jin ChungSo Yon AnTae Sung HwangSoo Yong LeeJeong-Su KimNam-Chul HaGyu-Yong SongBum-Joon Park Progerinin, an optimized progerin-lamin A binding inhibitor, ameliorates premature senescence phenotypes of Hutchinson-Gilford progeria syndrome. Communications Biology 2021, 4, 1-11, 10.1038/s42003-020-01540-w.
- Luke W. Koblan; Michael R. Erdos; Christopher Wilson; Wayne A. Cabral; Jonathan M. Levy; Zheng-Mei Xiong; Urraca L. Tavarez; Lindsay M. Davison; Yantenew G. Gete; Xiaojing Mao; et al.Gregory A. NewbySean P. DohertyNarisu NarisuQuanhu ShengChad KrilowCharles Y. LinLeslie B. GordonKan CaoFrancis S. CollinsJonathan D. BrownDavid R. Liu In vivo base editing rescues Hutchinson–Gilford progeria syndrome in mice. Nature 2021, 589, 608-614, 10.1038/s41586-020-03086-7.
- Xue Chen; Haidong Yao; Muhammad Kashif; Gwladys Revêchon; Maria Eriksson; Jianjiang Hu; Ting Wang; Yiran Liu; Elin Tüksammel; Staffan Strömblad; et al.Ian M AhearnMark R PhilipsClotilde WielMohamed X IbrahimMartin O Bergo A small-molecule ICMT inhibitor delays senescence of Hutchinson-Gilford progeria syndrome cells. eLife 2021, 10, e63284, 10.7554/elife.63284.
- Sakthivel, K.M.; Sehgal, P. A Novel Role of Lamins from Genetic Disease to Cancer Biomarkers. Oncol. Rev. 2016, 10, 309.
- Lochs, S.; Kefalopoulou, S.; Kind, J. Lamina Associated Domains and Gene Regulation in Development and Cancer. Cells 2019, 8, 271.
- Foster, C.R.; Robson, J.L.; Simon, W.J.; Twigg, J.; Cruikshank, D.; Wilson, R.G.; Hutchison, C.J. The role of Lamin A in cytoskeleton organization in colorectal cancer cells: A proteomic investigation. Nucleus 2011, 2, 434–443.
- Moss, S.F.; Krivosheyev, V.; de Souza, A.; Chin, K.; Gaetz, H.P.; Chaudhary, N.; Worman, H.J.; Holt, P.R. Decreased and aberrant nuclear lamin expression in gastrointestinal tract neoplasms. Gut 1999, 45, 723–729.
- Foster, C.R.; Przyborski, S.A.; Wilson, R.G.; Hutchison, C.J. Lamins as cancer biomarkers. Biochem. Soc. Trans. 2010, 38, 297–300.
- Contu, F.; Rangel-Pozzo, A.; Trokajlo, P.; Wark, L.; Klewes, L.; Johnson, N.A.; Petrogiannis-Haliotis, T.; Gartner, J.G.; Garini, Y.; Vanni, R.; et al. Distinct 3D Structural Patterns of Lamin A/C Expression in Hodgkin and Reed-Sternberg Cells. Cancers 2018, 10, 286.
- Wang, A.S.; Kozlov, S.V.; Stewart, C.L.; Horn, H.F. Tissue specific loss of A-type lamins in the gastrointestinal epithelium can enhance polyp size. Differentiation 2015, 89, 11–21.
- Gong, G.; Chen, P.; Li, L.; Tan, H.; Zhou, J.; Zhou, Y.; Yang, X.; Wu, X. Loss of lamin A but not lamin C expression in epithelial ovarian cancer cells is associated with metastasis and poor prognosis. Pathol. Res. Pract. 2015, 211, 175–182.
- Aljada, A.; Doria, J.; Saleh, A.M.; Al-Matar, S.H.; AlGabbani, S.; Shamsa, H.B.; Al-Bawab, A.; Ahmed, A.A. Altered Lamin A/C splice variant expression as a possible diagnostic marker in breast cancer. Cell. Oncol. 2016, 39, 161–174.
- Kaspi, E.; Frankel, D.; Guinde, J.; Perrin, S.; Laroumagne, S.; Robaglia-Schlupp, A.; Ostacolo, K.; Harhouri, K.; Tazi-Mezalek, R.; Micallef, J.; et al. Low lamin A expression in lung adenocarcinoma cells from pleural effusions is a pejorative factor associated with high number of metastatic sites and poor Performance status. PLoS ONE 2017, 12, e0183136.
- Zhang, Y.; Wang, J.; Huang, W.; Cai, J.; Ba, J.; Wang, Y.; Ke, Q.; Huang, Y.; Liu, X.; Qiu, Y.; et al. Nuclear Nestin deficiency drives tumor senescence via lamin A/C-dependent nuclear deformation. Nat. Commun. 2018, 9, 3613.
- Moiseeva, O.; Lessard, F.; Acevedo-Aquino, M.; Vernier, M.; Tsantrizos, Y.S.; Ferbeyre, G. Mutant lamin A links prophase to a p53 independent senescence program. Cell Cycle 2015, 14, 2408–2421.
- Mitchell, M.J.; Denais, C.; Chan, M.F.; Wang, Z.; Lammerding, J.; King, M.R. Lamin A/C deficiency reduces circulating tumor cell resistance to fluid shear stress. Am. J. Physiol. Cell Physiol. 2015, 309, C736–C746.
- Willis, N.D.; Cox, T.R.; Rahman-Casañs, S.F.; Smits, K.; Przyborski, S.A.; van den Brandt, P.; van Engeland, M.; Weijenberg, M.; Wilson, R.G.; de Bruïne, A.; et al. Lamin A/C is a risk biomarker in colorectal cancer. PLoS ONE 2008, 3, e2988.
- Wang, Y.; Jiang, J.; He, L.; Gong, G.; Wu, X. Effect of lamin-A expression on migration and nuclear stability of ovarian cancer cells. Gynecol. Oncol. 2019, 152, 166–176.
- Reis-Sobreiro, M.; Chen, J.F.; Novitskaya, T.; You, S.; Morley, S.; Steadman, K.; Gill, N.K.; Eskaros, A.; Rotinen, M.; Chu, C.Y.; et al. Emerin Deregulation Links Nuclear Shape Instability to Metastatic Potential. Cancer Res. 2018, 78, 6086–6097.
- Nardella, M.; Guglielmi, L.; Musa, C.; Iannetti, I.; Maresca, G.; Amendola, D.; Porru, M.; Carico, E.; Sessa, G.; Camerlingo, R.; et al. Down-regulation of the Lamin A/C in neuroblastoma triggers the expansion of tumor initiating cells. Oncotarget 2015, 6, 32821–32840.
- Zhang, X.; Lv, Y. Suspension state increases reattachment of breast cancer cells by up-regulating lamin A/C. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2272–2282.
- Kong, L.; Schäfer, G.; Bu, H.; Zhang, Y.; Zhang, Y.; Klocker, H. Lamin A/C protein is overexpressed in tissue-invading prostate cancer and promotes prostate cancer cell growth, migration and invasion through the PI3K/AKT/PTEN pathway. Carcinogenesis 2012, 33, 751–759.
- Zuo, L.; Zhao, H.; Yang, R.; Wang, L.; Ma, H.; Xu, X.; Zhou, P.; Kong, L. Lamin A/C might be involved in the EMT signalling pathway. Gene 2018, 663, 51–64.
- Capo-chichi, C.D.; Cai, K.Q.; Smedberg, J.; Ganjei-Azar, P.; Godwin, A.K.; Xu, X.X. Loss of A-type lamin expression compromises nuclear envelope integrity in breast cancer. Chin. J. Cancer 2011, 30, 415–425.
- Matsumoto, A.; Hieda, M.; Yokoyama, Y.; Nishioka, Y.; Yoshidome, K.; Tsujimoto, M.; Matsuura, N. Global loss of a nuclear lamina component, lamin A/C, and LINC complex components SUN1, SUN2, and nesprin-2 in breast cancer. Cancer Med. 2015, 4, 1547–1557.
- Wang, J.; Kondo, T.; Nakazawa, T.; Oishi, N.; Mochizuki, K.; Katoh, R. Constitutional abnormality of nuclear membrane proteins in small cell lung carcinoma. Virchows Arch. 2019, 475, 407–414.
- Wazir, U.; Ahmed, M.H.; Bridger, J.M.; Harvey, A.; Jiang, W.G.; Sharma, A.K.; Mokbel, K. The clinicopathological significance of lamin A/C, lamin B1 and lamin B receptor mRNA expression in human breast cancer. Cell. Mol. Biol. Lett. 2013, 18, 595–611.
- Wu, Z.; Wu, L.; Weng, D.; Xu, D.; Geng, J.; Zhao, F. Reduced expression of lamin A/C correlates with poor histological differentiation and prognosis in primary gastric carcinoma. J. Exp. Clin. Cancer Res. 2009, 28, 8.
- Cicchillitti, L.; Corrado, G.; Carosi, M.; Dabrowska, M.E.; Loria, R.; Falcioni, R.; Cutillo, G.; Piaggio, G.; Vizza, E. Prognostic role of NF-YA splicing isoforms and Lamin A status in low grade endometrial cancer. Oncotarget 2017, 8, 7935–7945.
- Capo-chichi, C.D.; Aguida, B.; Chabi, N.W.; Cai, Q.K.; Offrin, G.; Agossou, V.K.; Sanni, A.; Xu, X.X. Lamin A/C deficiency is an independent risk factor for cervical cancer. Cell. Oncol. 2016, 39, 59–68.
- Belt, E.J.; Fijneman, R.J.; van den Berg, E.G.; Bril, H.; Delis-van Diemen, P.M.; Tijssen, M.; van Essen, H.F.; de Lange-de Klerk, E.S.; Beliën, J.A.; Stockmann, H.B.; et al. Loss of lamin A/C expression in stage II and III colon cancer is associated with disease recurrence. Eur. J. Cancer 2011, 47, 1837–1845.
- Skvortsov, S.; Schäfer, G.; Stasyk, T.; Fuchsberger, C.; Bonn, G.K.; Bartsch, G.; Klocker, H.; Huber, L.A. Proteomics profiling of microdissected low- and high-grade prostate tumors identifies Lamin A as a discriminatory biomarker. Proteome Res. 2011, 10, 259–268.
- Jansen, M.P.; Machiels, B.M.; Hopman, A.H.; Broers, J.L.; Bot, F.J.; Arends, J.W.; Ramaekers, F.C.; Schouten, H.C. Comparison of A and B-type lamin expression in reactive lymph nodes and nodular sclerosing Hodgkin’s disease. Histopathology 1997, 31, 304–312.
- Alhudiri, I.M.; Nolan, C.C.; Ellis, I.O.; Elzagheid, A.; Rakha, E.A.; Green, A.R.; Chapman, C.J. Expression of Lamin A/C in early-stage breast cancer and its prognostic value. Breast Cancer Res. Treat. 2019, 174, 661–668.

