 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ashley Sutherland | + 3843 word(s) | 3843 | 2020-10-07 07:37:36 | | | |
| 2 | Rita Xu | -1097 word(s) | 2746 | 2021-02-25 03:53:16 | | |
Video Upload Options
The engineering of T cells through expression of chimeric antigen receptors (CARs) against tumor-associated antigens (TAAs) has shown significant potential for use as an anti-cancer therapeutic. The development of strategies for flexible and modular CAR T systems is accelerating, allowing for multiple antigen targeting, precise programming, and adaptable solutions in the field of cellular immunotherapy. Moving beyond the fixed antigen specificity of traditional CAR T systems, the modular CAR T technology splits the T cell signaling domains and the targeting elements through use of a switch molecule. The activity of CAR T cells depends on the presence of the switch, offering dose-titratable response and precise control over CAR T cells.
1. Introduction
Engineering T cells to express chimeric antigen receptors (CARs) has shown wide-ranging potential as a potent anti-cancer therapeutic. Characteristically, CARs consist of an extracellular antigen-binding single-chain antibody variable fragment (scFv) and hinge region linked to transmembrane and intracellular signaling regions. This engineered construct fuses the specificity of an antibody to T cell-effector functions, allowing for target cell lysis, release of cytokines, and T cell proliferation [1][2]. In clinical trials, CAR T cell therapy has shown remarkable success in treating hematological malignancies by targeting B cell antigen CD19. Numerous studies showed high remission rates, rapid tumor eradication, and durable responses in patients with refractory disease, raising expectations for expanding the types of cancers that can be treated with CAR therapy [3][4][5]. Although these results are encouraging, several challenges exist that inhibit the broad application of this treatment. Firstly, tumor heterogeneity is a complication that hinders CAR T development. Conventional CAR T cells have a fixed, single-antigen targeting ability, making the therapy vulnerable to antigen-loss relapse due to downregulation or antigen deletion [6][7]. CAR T therapy targeting a single antigen may initially demonstrate tumor regression; however, many cases have been reported in clinical trials of antigen-negative relapse after CD19 CAR T therapy due to tumor antigen escape [8][9]. Engineering of CAR T cells against a variety of tumor-associated antigens (TAAs) is a method to overcome tumor immunoediting, yet this approach comes with its own set of challenges. Further clinical success of CARs would necessitate the engineering of T cells tailored for each patient targeting various TAAs. However, significant technical requirements and financial costs involved in the generation and optimization of CARs directed at individual antigens limit this approach’s usefulness. To circumvent the technical and economic challenges of individually manufacturing and testing each new CAR, creating a platform using ‘universal’ redirected T cells against virtually any cell surface antigen is of particular importance for the rapid screening in pre-clinical models and the broad application of CAR T therapy.
The ability to generate modular or universal CARs hinges on the separation of targeting and signaling elements. Modular CAR T cells are not targeted at the tumor antigen itself; instead, the CAR is directed at an adaptor or switch element (Figure 1). This adaptor serves as the targeting element, binding to the tumor antigen, and is required to bridge the immunological synapse. The firing of the CAR T should occur only in the presence of the switch, and swapping out the adaptor molecule allows for redirection of the T cell without the need for re-engineering and time-consuming remanufacturing. This modular treatment approach offers the possibility for flexibility in tumor targeting in the clinic; adapting with the patient’s tumor by adjusting treatment based on the cancer’s changing antigen expression could be envisioned. Fixed antigen targeting often hinders cancer treatment, with this modular CAR T approach driving an already innovative biological therapy into truly tailored cancer therapy.
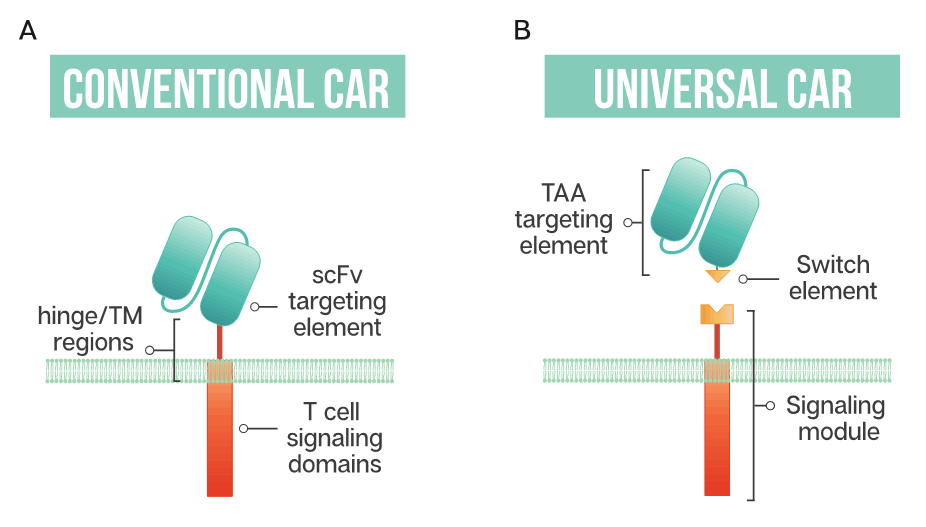
Figure 1. Schematic representation of a conventional CAR T cell and a universal or modular CAR T cell (A) Conventional CAR T cells have a single-chain antibody fragment (scFv) targeting element, expressed in tandem with signaling domains derived from the T cell receptor and costimulatory domains such as 4-1BB and CD28 connected through a transmembrane (TM) domain and a flexible spacer or hinge region; (B) a universal CAR T cell has a split design containing a tumor-associated antigen (TAA) targeting element, usually derived from a monoclonal antibody, a switch element and the signaling module, consisting of the T cell signaling domains and an extracellular region which interacts with the switch element.
In the toolkit of building modular CARs, the number of adaptors has been increasing, with components such as immunoglobulin (IgG)-based adaptors such as scFvs (single-chain variable fragment), Fabs (antigen-binding fragment), nanobodies, and full-length IgGs being the most established. Both antibody-based and targeting ligand adaptors have been redirected using tags attached either genetically or post-translationally and include peptide tags such as neo-epitopes, SpyTag, leucine zippers, biotin and fluorescein isothiocyanate (FITC). Repurposing clinically approved IgGs and adding redirecting tags could reduce the regulatory hurdles and allow for a suite of targeting elements that clinicians could employ for a wide variety of cancer indications.
Improving the safety profile of CAR T therapy is a rapidly developing area of research with the goal of expanding the therapy to treat a broader range of cancer patients. With the promise of CAR T therapy comes life-threatening side effects, including severe cytokine release syndrome (CRS), neurological toxicities and organ failure, resulting from the unrestrained proliferation of CAR T cells [10][11]. Toxicities related to on-target off-tumor reactions can occur when low levels of the targeted cancer antigen found on normal tissue are targeted by the engineered T cells. When toxicities become severe enough, administration of high-dose corticosteroids is required, decreasing the T cell numbers [10][11][12]. Research in the area of ‘next-generation’ CAR T cells has incorporated methods such as suicide genes or switches as a means to eliminate T cells. These ‘emergency stop’ methods may avert a lethal outcome to the patient; however, the drawback is that the CAR T cells are eliminated, and with it any therapeutic response—a bad ending for a costly and time-consuming therapy. Universal or modular CAR T strategies that require administration of an adaptor may fit the criteria for better control of engineered T cells. The ability to titrate on adaptors could facilitate the ‘turning off’ of CAR T cells by halting administration of the adaptor, possibly enhancing their safety profile without the need to destroy the T cells. Additionally, this tunable response could better manage side effects as well as fine tuning of CAR T activity. These approaches for CAR T generation allow for conjugation of the tagged targeting element with the anti-tag CAR T. This enables targeting multiple TAAs and flexibility in administration of the targeting element, a step towards overcoming current clinical limitations of CAR T therapy.
2. Modular CAR T Platforms
2.1. Biotin-Binding Immune Receptors
Exploiting one of nature’s strongest non-covalent bonds, the avidin–biotin interaction has been used to generate a universal tumor-targeting system, biotin-binding immunoreceptor (BBIR) (Figure 2A). BBIRs constructed by Urbanska et al. [13] consist of an extracellular modified dimeric avidin (dcAv) linked to an intracellular T cell signaling domain. This split design allows for easy target modification and targeting of multiple antigens. Monomeric avidin was unable to elicit an effector response, most likely due to the decreased affinity between biotin and monomeric avidin.
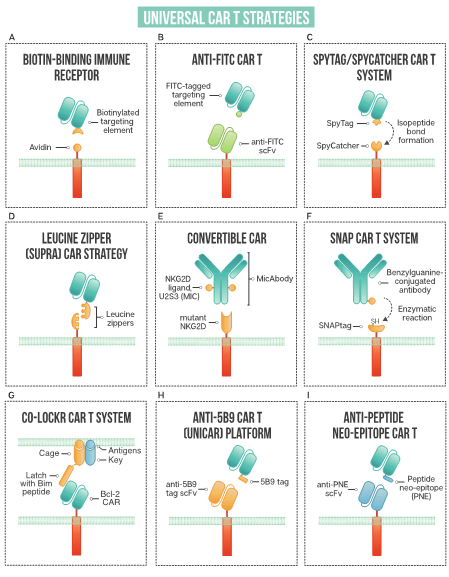
Figure 2. A schematic of strategies used in universal CAR T design: (A) biotin-binding immune receptor; (B) anti-FITC CAR T; (C) the SpyTag/SpyCatcher CAR T system; (D) leucine zipper or SUPRA CAR T; (E) convertibleCAR or modified NKG2D CAR T; (F) SNAP CAR T enzymatic CAR labeling system; (G) the co-localization-dependent protein switch (Co-LOCKR) CAR T system; (H) UniCAR or anti-5B9 peptide CAR platform; (I) anti-peptide neo-epitope (PNE) CAR T.
Tumor cells in vitro are either pre-targeted or co-administered with biotinylated antigen-targeting molecules (IgG antibodies, scFvs, or other tumor-specific ligands), and T cells expressing BBIRs bound specifically to exert effector cell functions. In vitro functioning of dcAv BBIR is comparable to traditional CAR T for cell lysis and cytokine secretion, illustrating this system’s utility to target cells and perform effector functions. Significantly, BBIR cells generated a dose-dependent response with the addition of adaptors. When tested against a panel of cells with variable TAAs, BBIRs could target antigens both simultaneously or sequentially, showing the tunability of CAR response and their utility in antigen escape scenarios. Furthermore, the authors proposed a platform using BBIRs to screen candidate antibodies or other targeting elements in vitro for rapid pre-clinical screening. BBIRs performed similarly in vivo in a xenograft mouse model of human ovarian cancer. As CAR safety is essential for moving into the clinic, BBIRs were exposed to supraphysiological levels of biotin and showed no antigen-independent activation. Interestingly, T cells could not be ‘pre-armed’ with the biotinylated targeting element; the adaptor had to be either pre-targeted to coat the tumor surface or co-administered with BBIR T cells.
Lohmueller et al. [14] drew on this system and further affinity enhanced streptavidin, designing a biotin-binding domain where monomeric streptavidin could be used in the CAR system with higher affinity to biotin than the dimeric form. This affinity-enhanced form, mSA2, has a more compact structure than the dimeric form employed previously, possibly increasing the type of antigens able to be targeted by this system. The mSA2 CARs were able to distinguish antigen-positive cells precoated with biotinylated antibody in vitro and produce a specific effector response. Antigen-negative tumor cells did not elicit an effector response, neither did non-binding biotinylated antibody, showing that in order for CAR T cells to be turned on, the antibody must be bound to the target cells. The mSA2 CAR T cells showed potent effector functions; however, its potential immunogenicity could hamper its adoption in the clinic. With the BBIR system, excess biotin does not impart an inhibitory effect, and therefore could not be used as a potential ‘off switch’ for added safety.
2.2. Anti-FITC CAR Strategy
Fluorescein isothiocyanate (FITC), derived from fluorescein, is a fluorescent label commonly used to tag antibodies. Tamada et al. [15] first exploited this common labeling method and generated anti-FITC CAR T cells able to be directed by FITC-tagged antibodies (Figure 2B). The extracellular portion of the CAR is comprised of an anti-FITC scFv that recognizes FITC-labeled cetuximab (anti-EGFR), trastuzumab (anti-HER2), and rituximab (anti-CD20), antibodies that are already employed clinically. Redirected CAR T cells were found to be effective both in vitro and in vivo to specifically bind their respective tumor cells and exert anti-tumor effects. This system showed effectiveness at targeting multiple TAAs to better address heterogenous cancer populations. Additionally, the use of anti-FITC CAR T cells was shown to restore the usefulness of monoclonal antibodies to additional cancer types. They discuss that in patients with Kras mutations, cetuximab does not provide therapeutic benefits; however, when cetuximab is utilized with anti-FITC CAR T cells, anti-tumor effects are shown, as illustrated with the SW480 cell line that containing a Kras mutation. The anti-FITC CAR T system was applied using trastuzumab—Cao et al. conjugated FITC to trastuzumab in a site-specific manner compared to another strategy where a peptide neo-epitope (PNE) was fused to trastuzumab [16]. Both antibody tagging methods showed a dose-titratable immune response, capable of completely clearing HER2-positive tumors in vivo. The first clinical use of trastuzumab incorporated into a CAR T resulted in a serious adverse event, with the patient developing on-target, off-tumor toxicity related to the redirection of CAR T cells to lung epithelium, proving fatal [17]. Since this initial trial, many groups have investigated safer ways to target HER2, reviewed by Liu et al. [18], with the modular anti-FITC CAR T technology, a contender to address the safety issues with targeting this cancer-associated antigen.
Expanding the targeting elements to more than full-length antibodies, Zhang et al. employed switchable CAR-engineered T cells using anti-tumor peptides that specifically target integrin avβ3 through an 18-amino acid sequence fused to FITC [19]. This peptide adaptor molecule, termed FITC-HM-3, specifically targeted tumor cells and regulated CAR T cell activity. Demonstrating that low-molecular-weight switch molecules can be effective at redirecting engineered T cells, Lee et al. [20] employed a cocktail of small bifunctional molecules in conjunction with anti-fluorescein CAR T cells to target cancer cells in vitro and in vivo. The bifunctional molecules, called CAR T cell adapter molecules (CAMs), consist of fluorescein linked to a tumor-specific ligand through a hydrophilic spacer. The use of a mixture of CAMs enables the targeting of heterogenous solid tumors and broadens the applicability of CAR T cell therapy by using small molecules, which could improve tumor penetration, as opposed to larger full-length antibodies. Additionally, improved safety is offered by the short half-life (~90 min) of small molecules, allowing them to rapidly clear from receptor-negative tissue.
Optimizing the complex between the CAR T cell, switch, and tumor antigen is essential for optimal CAR T activation and cell killing. Using the modular CAR system, Ma et al. [21] utilized anti-FITC CARs to target both CD19 and CD22, whereby antibody fragments were site-specifically modified with FITC through genetically encoded non-canonical amino acids. This allowed for the incorporation of FITC to optimize of the geometry of the immunological synapse. Compared head to head, the optimized anti-FITC CAR T targeting CD19 performed similarly to conventional CD19-targeting CAR T, necessary for moving this technology forward into the clinic. Furthermore, excess FITC at 10 μM was shown to dampen CAR T activity in vitro, a feature that could be used to improve safety in the clinic. Others have shown that the addition of FITC-labeled non-specific antibodies could also be used to attenuate CAR T cells [15].
The targeting of folate receptors using anti-FITC CARs has been demonstrated by several groups [22][23][24]. Lu et al. [23], using FITC conjugated to folic acid as the switch molecule, modeled severe cytokine release syndrome and determined that CRS could be alleviated through the titration of the folate FITC adaptor or by intermittent dosing. Reversal of severe CRS could be achieved by intravenous sodium fluorescein to transiently interrupt CARs, without destroying the engineered T cells. With the ability to shut down the CAR T response through the addition of FITC [21], FITC labeled non-specific antibodies [15] or sodium fluorescein [23][24], this system with its added ‘safety switches’ could allow for engineered immune cell deactivation if toxicity develops, possibly being able to salvage the therapy by re-administering the switch molecules. While encouraging, the possible immunogenicity of FITC adaptors in the context of CAR T systems requires further study.
2.3. The SpyTag-SpyCatcher Universal CAR T System
The SpyTag/SpyCatcher protein ligation system employs a unique peptide: protein ligation reaction to link the tagged targeting element to the immune receptor. In 2012, Zackeri et al. reported a fibronectin-binding protein from Streptococcus pyogenes that, upon splitting it into two parts, followed by rational engineering, an N-terminal protein fragment (SpyCatcher) and a C-terminal 13-amino acid peptide (SpyTag) were produced [25]. The two parts will spontaneously reconstitute to form an isopeptide bond without the need for co-factors, enzymes, or specific conditions. Minutolo et al. exploited this system to generate a SpyCatcher immune receptor [26] (Figure 2C). This immune receptor contains the SpyCatcher protein as the extracellular domain, linked to intracellular signaling regions. TAA-specific targeting ligands, such as IgG antibodies, are site-specifically labeled with SpyTag. This post-translational covalent assembly allows for the redirection of T cells to multiple TAAs to exert targeted effector cell functions. The SpyCatcher immune receptor activity depends on the presence of both target antigen and SpyTag-labeled targeting element, allowing for titratable control of the engineered T cells. Arming the SpyCatcher CAR T cells with SpyTagged antibodies showed receptor levels decreasing over time, with complete loss observed after 96 h. SpyCatcher CAR T cells were shown to become functional upon the addition of SpyTag targeting element and lyse antigen-expressing target cells in vitro. Using an immunodeficient mouse xenograft model, the authors showed that HER2-positive xenografts could be targeted with SpyCatcher CAR T cells pre-armed with SpyTagged Herceptin. Additional targeting ligand was administered every three days, and administration throughout treatment was shown to be necessary for tumor clearance. The SpyTag/SpyCatcher system was tested using targeting elements against HER2, EGFR, EpCAM and CD20 and Liu et al. expanded the range of targetable antigens by demonstrating SpyCatcher immune receptors could be constructed to target the hepatocellular carcinoma antigen, human glypican-3 (hGPC3) using a SpyTagged anti-hGPC3 scFv [27].
Potential immunogenicity is an issue that may hamper SpyCatcher immune receptor adoption in the clinic. Owing to its bacterial origin, the Tag/Catcher system may be vulnerable to recognition by the patient’s immune system. Work has been performed in developing SpyCatcher/SpyTag variants with truncations aimed at reducing potential immunogenicity [28] and tested using immunocompetent mice, but further study is needed to determine the likelihood of adverse reactions in humans.
References
- Sadelain, ; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398, doi:10.1158/2159-8290.CD-12-0548.
- Shirasu, ; Kuroki, M. Functional Design of Chimeric T-Cell Antigen Receptors for Adoptive Immunotherapy of Cancer: Architecture and Outcomes. Anticancer. Res. 2012, 32, 2377–2383.
- Brentjens, J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38, doi:10.1126/scitranslmed.3005930.
- Lee, W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528, doi:10.1016/S0140-6736(14)61403-3.
- Kochenderfer, N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood 2012, 119, 2709–2720, doi:10.1182/blood-2011-10-384388.
- Xu, ; Sun, Q.; Liang, X.; Chen, Z.; Zhang, X.; Zhou, X.; Li, M.; Tu, H.; Liu, Y.; Tu, S.; et al. Mechanisms of Relapse After CD19 CAR T-Cell Therapy for Acute Lymphoblastic Leukemia and Its Prevention and Treatment Strategies. Front. Immunol. 2019, 10, 2664, doi:10.3389/fimmu.2019.02664.
- Kailayangiri, ; Altvater, B.; Wiebel, M.; Jamitzky, S.; Rossig, C. Overcoming Heterogeneity of Antigen Expression for Effective CAR T Cell Targeting of Cancers. Cancers 2020, 12, 1075, doi:10.3390/cancers12051075.
- Maude, L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448, doi:10.1056/NEJMoa1709866.
- Park, H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459, doi:10.1056/NEJMoa1709919.
- Chen, ; Wang, F.; Zhang, P.; Zhang, Y.; Chen, Y.; Fan, X.; Cao, X.; Liu, J.; Yang, Y.; Wang, B.; et al. Management of cytokine release syndrome related to CAR-T cell therapy. Front. Med. 2019, 13, 610–617, doi:10.1007/s11684-019-0714-8.
- Brudno, N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55, doi:10.1016/j.blre.2018.11.002.
- Thakar, S.; Kearl, T.J.; Malarkannan, S. Controlling Cytokine Release Syndrome to Harness the Full Potential of CAR-Based Cellular Therapy. Front. Oncol. 2020, 9, 1529, doi:10.3389/fonc.2019.01529.
- Urbanska, ; Lanitis, E.; Poussin, M.; Lynn, R.C.; Gavin, B.P.; Kelderman, S.; Yu, J.; Scholler, N.; Powell, D.J. A Universal Strategy for Adoptive Immunotherapy of Cancer through Use of a Novel T-cell Antigen Receptor. Cancer Res. 2012, 72, 1844–1852, doi:10.1158/0008-5472.CAN-11-3890.
- Lohmueller, J.; Ham, J.D.; Kvorjak, M.; Finn, O.J. mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. OncoImmunology 2018, 7, e1368604, doi:10.1080/2162402X.2017.1368604.
- Tamada, ; Geng, D.; Sakoda, Y.; Bansal, N.; Srivastava, R.; Li, Z.; Davila, E. Redirecting Gene-Modified T Cells toward Various Cancer Types Using Tagged Antibodies. Clin. Cancer Res. 2012, 18, 6436–6445, doi:10.1158/1078-0432.CCR-12-1449.
- Cao, ; Rodgers, D.T.; Du, J.; Ahmad, I.; Hampton, E.N.; Ma, J.S.Y.; Mazagova, M.; Choi, S.; Yun, H.Y.; Xiao, H.; et al. Design of Switchable Chimeric Antigen Receptor T Cells Targeting Breast Cancer. Angew. Chem. Int. Ed. 2016, 55, 7520–7524, doi:10.1002/anie.201601902.
- Morgan, A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851, doi:10.1038/mt.2010.24.
- Liu, ; Zhang, N.; Shi, H. Driving better and safer HER2-specific CARs for cancer therapy. Oncotarget 2017, 8, 62730–62741, doi:10.18632/oncotarget.17528.
- Zhang, ; Gu, J.; Xue, J.; Lin, C.; Liu, C.; Li, M.; Hao, J.; Setrerrahmane, S.; Chi, X.; Qi, W.; et al. Accurate control of dual-receptor-engineered T cell activity through a bifunctional anti-angiogenic peptide. J. Hematol. Oncol. 2018, 11, 44, doi:10.1186/s13045-018-0591-7.
- Lee, G.; Marks, I.; Srinivasarao, M.; Kanduluru, A.K.; Mahalingam, S.M.; Liu, X.; Chu, H.; Low, P.S. Use of a Single CAR T Cell and Several Bispecific Adapters Facilitates Eradication of Multiple Antigenically Different Solid Tumors. Cancer Res. 2019, 79, 387–396, doi:10.1158/0008-5472.CAN-18-1834.
- Ma, S.Y.; Kim, J.Y.; Kazane, S.A.; Choi, S.-H.; Yun, H.Y.; Kim, M.S.; Rodgers, D.T.; Pugh, H.M.; Singer, O.; Sun, S.B.; et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E450–E458, doi:10.1073/pnas.1524193113.
- Chu, ; Zhou, Y.; Tang, Q.; Wang, M.; Ji, Y.; Yan, J.; Yin, D.; Zhang, S.; Lu, H.; Shen, J. Bi-specific ligand-controlled chimeric antigen receptor T-cell therapy for non-small cell lung cancer. Biosci. Trends 2018, 12, 298–308, doi:10.5582/bst.2018.01048.
- Lu, J.; Chu, H.; Wheeler, L.W.; Nelson, M.; Westrick, E.; Matthaei, J.F.; Cardle, I.I.; Johnson, A.; Gustafson, J.; Parker, N.; et al. Preclinical Evaluation of Bispecific Adaptor Molecule Controlled Folate Receptor CAR-T Cell Therapy With Special Focus on Pediatric Malignancies. Front. Oncol. 2019, 9, 151, doi:10.3389/fonc.2019.00151.
- Kim, S.; Ma, J.S.Y.; Yun, H.; Cao, Y.; Kim, J.Y.; Chi, V.; Wang, D.; Woods, A.; Sherwood, L.; Caballero, D.; et al. Redirection of Genetically Engineered CAR-T Cells Using Bifunctional Small Molecules. J. Am. Chem. Soc. 2015, 137, 2832–2835, doi:10.1021/jacs.5b00106.
- Zakeri, ; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697, doi:10.1073/pnas.1115485109.
- Minutolo, G.; Sharma, P.; Poussin, M.; Shaw, L.C.; Brown, D.P.; Hollander, E.E.; Smole, A.; Rodriguez-Garcia, A.; Hui, J.Z.; Zappala, F.; et al. Quantitative Control of Gene-Engineered T-Cell Activity through the Covalent Attachment of Targeting Ligands to a Universal Immune Receptor. J. Am. Chem. Soc. 2020, 142, 6554–6568, doi:10.1021/jacs.9b11622.
- Liu, ; Wen, J.; Yi, H.; Hou, X.; Yin, Y.; Ye, G.; Wu, X.; Jiang, X. Split chimeric antigen receptor-modified T cells targeting glypican-3 suppress hepatocellular carcinoma growth with reduced cytokine release. Ther. Adv. Med. Oncol. 2020, 12, 1-16, doi:10.1177/1758835920910347.
- Liu, Z.; Zhou, H.; Wang, W.; Tan, W.; Fu, Y.-X.; Zhu, M. A novel method for synthetic vaccine construction based on protein assembly. Sci. Rep. 2014, 4, 7266, doi:10.1038/srep07266.

