 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Elisabetta Benedetti | + 4377 word(s) | 4377 | 2021-01-28 07:54:21 | | | |
| 2 | Rita Xu | -1760 word(s) | 2617 | 2021-02-23 09:35:10 | | |
Video Upload Options
The pathophysiological processes of inflammatory bowel diseases (IBDs), i.e., Crohn’s disease (CD) and ulcerative colitis (UC), are still not completely understood.
1. Introduction
The worldwide epidemiology of inflammatory bowel diseases (IBDs), such as Crohn’s disease (CD) and ulcerative colitis (UC), has been influenced mainly by industrial progress and the improvement of human living conditions [1]. The increasing incidence and prevalence of IBD in developing countries suggest a connection with a westernized lifestyle and changed habits. The etiopathogenesis of IBD is not entirely understood. However, it is hypothesized to be related to a mixture of factors, including genetic susceptibility, dysregulation of the gut immune system, and environmental elements in conjunction with the microbiota. There is a need for complete information about these diseases since they represent an expanding global health problem, the costs of which are challenging to manage.
CD and UC are chronic inflammatory disorders with distinct clinical characteristics. CD can affect all gastrointestinal tract segments (most commonly the terminal ileum and colon) in a non-continuous manner, causing a typically asymmetrical, segmental, and transmural inflammation. CD complications include abscesses, fistulas, and strictures, and many patients need surgical procedures. UC involves the colonic mucosal surface, primarily affecting the rectum and, in some cases, the entire colon in a continuous manner. Depending on the extent of inflammation, UC can evolve into several forms, from proctitis to left-sides colitis or pancolitis [2]. Behind the different characteristics, CD and UC share almost common features regarding risk factors, symptoms, clinical course, complications, and the absence of a definitive cure. The current therapeutic approaches aim to reduce intestinal inflammation and, more specifically, block pro-inflammatory cytokines. However, the conventional therapy of IBD, i.e., salicylates, steroids, and immunosuppressants reduce the mortality, but not the rate of complications and surgery; the latter is required in up to 70% of CD and 20% of UC patients during their lifetime. The introduction of biological therapies, i.e., anti-tumor necrosis factor-alpha (TNF-α), anti-α4β7 integrin, and anti-interleukins 12/23 antibodies, caused a revolution in the treatment of IBD; however, they reduced hospitalization, complications, and surgery only in the short- and medium-term. For these reasons, it is necessary to find personalized treatments employing new strategies for drug monitoring and, above all, identifying useful targets.
Concerning the underlying mechanism, IBD’s pathogenesis seems to be related to an alteration in the innate immune system, including epithelial barrier defects with changes in E-cadherin, β-catenin, and claudins expression [3] and an inadequate expression of antimicrobial peptides. The gut immune homeostasis can be disrupted by innate immune cells contribution (neutrophils, dendritic cells, and macrophages) and the release of inflammatory mediators [4]. IBDs are typically characterized by high levels of cytokines, such as TNF-α, IL-6, IL-8 (one of the first chemokines described), IL-12, and chemokines such as chemokine ligand 2, chemokine ligand 3, and chemokine ligand 1 in colon tissues [5]. However, the role of adaptive immunity cannot be omitted since, in CD and UC patients, an alteration in immunoglobulin subclass production has been found [6]. Among other pathogenic components, there is an impairment in Peroxisome proliferator-activated receptors-γ (PPARγ) activity, abnormalities of the enteric nervous system, genetic variants, and the presence of regulatory RNAs [4]. The high expression of PPARγ in the bowel has already been demonstrated [7], and several studies have shown its role in human colonic inflammation [8] along with the involvement of immune system response [9]. Notably, mesalazine, the most used drug in UC, binds and activates PPARγ [10]. Specifically, mesalazine enhances PPARγ expression and promotes its translocation from the cytoplasm to the nucleus [10]. Different xenobiotics and environmental pollutants can influence and alter the Peroxisome proliferator-activated receptors (PPAR) signaling pathway [11]. This evidence suggests a relationship between external factors and the onset of gut inflammation diseases.
2. PPARs: Crosstalk between Metabolism and Inflammation
PPARs are ligand-dependent transcription factors and belong to nuclear hormone receptors’ superfamily, playing an important role in lipid and glucose metabolism [12]. In mammals, three isoforms of PPARs have been identified: PPARα or NR1C1, PPARβ/δ or NR1C2, and PPARγ or NR1C, each of them encoded by a different gene. They share a similar structure. The ability to bind agonists is mediated by a ligand-binding domain (LBD) in the C-terminus, while the DNA binding domain is in the N-terminus. These receptors can be activated by natural fatty acids and eicosanoids or synthetic ligands, which are used in the clinical management of metabolic diseases, such as fibrates, with cardioprotective properties [13], and thiazolidinediones, used in the treatment of diabetes mellitus type 2 [14]. After interaction with agonists, they are translocated in the nucleus, and their function depends on the heterodimerization with retinoid X receptor (RXR). The heterodimers bind to sequence-specific PPAR response elements (PPREs), stimulating the target genes’ transcription [15].
The members of the PPARs family show a wide range of actions on glucose and lipidic homeostasis, and they share many similarities in terms of structure and function; however, each isoform has a specific physiological activity, influenced by their tissue distribution.
PPARα is expressed in tissues that require a large amount of energy, principally the liver, kidney, and skeletal muscle; its localization has also been demonstrated in cardiomyocytes, intestinal mucosa, adrenal gland, brown adipose tissue, and brain [16][17]. It is involved in the catabolism of fatty acids and their oxidation [18]. Regarding PPARβ/δ, this isoform is involved in several processes, including cell proliferation, differentiation, migration, and apoptosis. Its activity is also related to glucose and cholesterol homeostasis, insulin sensitivity, and angiogenesis [17][19]. It is ubiquitously expressed but particularly abundant in the gastrointestinal tract, kidneys, skeletal muscle, and brain [20]. PPARγ is abundantly expressed in white and brown adipose tissue, where it plays a crucial role in regulating adipogenesis, energy balance, and lipid biosynthesis. It is also expressed in the intestines, liver, kidneys, brain, immunological system, and muscles [17][21].
PPARs are usually described as the main actors in lipid and glucose metabolism, but much evidence indicates their involvement in controlling inflammatory responses and inflammation-related disorders such as fibrosis and cancer. In addition to their known anti-inflammatory action, PPARs also modulate fibrogenesis and carcinogenesis.
It has been reported that PPARs have anti-inflammatory potential, modulating several points of inflammatory pathways. Inflammation consists of a dynamic sequence of phenomena, including the release of mediators that leads to vasodilatation, increased blood flow, vascular permeability, and recruitment of polymorphonuclear cells, particularly neutrophils in acute phase of inflammatory process, whereas mononuclear cells, macrophages, T- and B-lymphocytes are in chronic immunomediated inflammation. PPARs could intervene at each level of these processes. For example, PPARα can negatively interfere with the NF-κB signaling pathway, repressing several inflammatory genes such as VCAM-1, COX-2, and IL-6 [22]. PPARα is also involved in inhibiting the expression of inducible nitric oxide synthase [23] and TNF-α in macrophages [24]. PPARβ/δ activity is induced in the host inflammatory response in the skin, and it results in being up-regulated in keratinocytes as a consequence of external triggers. The activation of PPAR-β/δ-pathway determines the expression of genes related to keratinocyte differentiation, survival, and repair [25]. Other studies focused on the role of PPARβ/δ in attenuating atherosclerosis progression, revealing that this isoform has an HDL-raising effect and anti-inflammatory activity within the vessel wall, where it participates in the down-regulation of chemokines production [26].
Mechanisms of the anti-inflammatory effects of PPARγ include the inhibition of the transcriptional activity of NF-κB, STAT-1, and AP-1 [27]. A direct relationship between TNF-α adipocyte secretion and a decrease in expression of PPARγ has been reported [28]. Moreover, negative regulation of PPARγ contributes to the antiadipogenic effects of TNF-α, whose increased production is relevant in obesity states [29]. In fact, several studies have also demonstrated a metabolic benefit related to the anti-inflammatory effects of targeting PPARγ [30].
All PPARs isoforms can have a role in regulating inflammatory responses, employing their interaction with various transcription factors stimulating inflammation, signal transducer, the formation of complexes between co-activators and co-repressors, and the modulation of different kinases [31].
Thus, the need for more in-depth knowledge of PPARs activity derives from their key role in various metabolic processes, including lipid and glucose homeostasis and inflammatory disease, which makes them the ideal target for developing new pharmacological strategies.
PPAR agonists are currently used to treat many diseases, such as hyperlipidemia, insulin resistance, type 2 diabetes, cardiometabolic syndrome, and atherosclerosis. The latest generation of agonists is represented by the selective peroxisome proliferator-activated receptor modulators, indicated as SPPARMs. With respect to traditional ligands, they can act as dual partial agonists, binding to two isoforms of receptors. Moreover, they have the ability to produce particular conformational changes, which leads to the preferential activation of transcriptional factors [32]. Further progress is represented by pan agonists, whose beneficial effects result from the ability to bind and activate the diverse isoforms of PPARs, reinforcing the single activation mediated by selective agonists [33].
The Importance of PPARγ in IBDs
Maintaining a healthy gastrointestinal tract depends on external factors (diet, chemicals, drugs, stress) or endogenous factors (genetics, microbiota, efficient immune responses). The loss of equilibrium between these factors may determine the onset of diseases. However, the elements contributing to inflammation and bowel disease are not clear. It is not possible to define a single factor or gene responsible for these modifications. Considering the critical role of PPARs in inflammation, many studies have focused on the possible correlation between these receptors, IBD, and cancer. In particular, chronic intestinal inflammation represents the leading risk factor for the development of gastrointestinal malignancy. Patients with UC and CD have a significantly higher cancer susceptibility [34].
The role of PPARs in inflammatory responses, which has already been described above, makes them possible actors in the pathogenesis of intestinal disorders and cancer. Particular attention has been given to PPARγ since it is abundantly expressed in epithelial cells in the large and small intestines [7]. Although the first evidence on the potential link between PPARγ and intestinal disease established a correlation between this receptor expression and an increase in colon tumorigenesis, recently, many researchers have re-examined the association between PPARγ and the risk of colorectal cancer (CRC) [35][36][37][38].
Genetic studies have demonstrated that heterozygous intestinal-specific PPARγ knockout enhanced tumor growth, evidencing PPARγ as a tumor resistance factor [39].
Interestingly, the activation of PPARγ by mesalazine could be responsible for CRC prevention observed with this drug in IBDs [40]. In immune-deficient mice engrafted with human CRC cells, mesalazine administration reduces xenografts’ growth via a PPAR-γ-dependent mechanism [41]. Activation of PPARγ by mesalazine is accompanied by induction of the tumor suppressor gene PTEN, activation of caspase-8 and caspase-3, and diminished expression of surviving and X-linked inhibitor apoptosis protein [42].
However, the role of PPARγ is not only related to tumorigenesis, because a lot of evidence suggests its involvement in inflammation diseases. For example, a down-regulation has been reported in PPARγ expression in UC [37], and a negative correlation has also been hypothesized with UC progression, because of the low level of PPARγ mRNA in the mucosa of active UC patients compared with UC patients in remission [8]. Sugawara et al. demonstrated positive evidence for the association of allelic variation of the PPARγ gene and CD [43]. These findings suggest that chronic inflammation could be caused by decreased levels in PPARγ expression in the colon.
Among the distinctive features of IBDs’ pathogenesis, there is real deregulation in cytokine production in inflamed colon areas. Studies on human biopsies and in vitro models have demonstrated that a broad set of molecules dominates the mucosal response, where the contribution of epithelial cells is predominant [44]. These soluble mediators include pro-inflammatory cytokines, such as TNF-α, IFN-γ, IL-6, IL-12, IL-21, IL-23, IL-17, and anti-inflammatory cytokines, such as IL-10, TGFβ, IL-35 [4] and chemokines CXCL1, CXCL2, CXCL3, CCL20 [44]. In particular, the elevated production of IL-12, IL-23, IFN-γ, and IL-17 seems to be characteristic of CD, while UC is usually associated with increased production of IL-3, IL-5, and IL-9 [5].
PPARγ distinguishes itself for the protective effects, including the modulation of cytokine/chemokine production and the negative regulation of macrophage activation [45]. In this scenario, it is not difficult to understand why studies have abundantly focused on the γ-isoform of PPARs and the possible mechanism through which this receptor could be involved in bowel inflammation (Figure 1).
The activation of PPARγ determines a decrease in the production of pro-inflammatory cytokines, such as TNF-α and IL-6, and the inhibition of transcription factors, including NF-κB, AP-1, STAT-1, and Intercellular Adhesion Molecule (ICAM-1), and matrix metallopeptidase 9 (MMP-9) [46].
Several studies have shown that PPARγ synthetic agonists can ameliorate gut inflammatory phenomena. Bassaganya-Riera et al. [47], using in vivo models of IBD, provided molecular evidence that conjugated linoleic acid reduces colitis inflammation employing a PPAR γ-dependent mechanism.
More recently, it was shown that the synthesized jasmonate analog J11-Cl (2-hydroxyethyl 5-chloro-4,5-didehydrojasmonate), structurally similar to cyclopentenone prostaglandin 15d-PGJ2, increases PPARγ activity and exerts anti-inflammatory effects, determining a less severe form of intestinal inflammation in dextran sodium sulfate (DSS)-induced colitis in mice. The treatment reduced pro-inflammatory cytokines and chemokines and increased anti-inflammatory cytokines and growth factors [48]. Another research study also supports the role of PPARγ in the amelioration of inflammatory bowel disease. In trinitrobenzene sulfonic acid (TNBS)-induced colitis mice, the andrographolide-lipoic acid conjugate (AL-1) administration alleviates inflammation through inhibiting the expression of TNF-α, IL-1β, and IL-6 and down-regulating the expression of p65 and p-IκB, key regulators of NF-κB pathway. Moreover, COX-2 levels, which are regulated by NF-κB, were reported to control levels, and the expression of PPARγ was increased in AL-1 treated groups [49]. The modulation of PPARγ/NF-κB cascade in intestinal inflammation is related to p21-activated kinase 1 (PAK1), the results of which overexpressed and activated in IBDs [50]. In particular, TNF-α is responsible for the translocation and co-localization of p-PAK1 and p-65 in the nucleus. These events determine the transcriptional activation of NF-κB. Activated PAK1 blocks PPARγ, increasing accumulation of p-65.
TNF-α stimulation can also induce the expression of COX-2, whose transcription can be regulated by several factors such as NF-κB and PPARs [51]. It has been demonstrated that this pro-inflammatory enzyme is induced in the human inflamed large intestine and IL-10 deficient mouse model of IBD [52][53]. COX-2 metabolizes free arachidonic acid (AA) into prostanoids, such as prostaglandins (PGs) and thromboxanes (TXs). The by-products cyclopentenone prostanoids, including 15-deoxy-12,13-didehydro-14,15-didehydro-PGJ2 (15d-Δ12,14-PGJ2), 12,13-didehydro-PGJ2 (Δ12-PGJ2), and PGA2, are ligands of PPARγ, suggesting an interaction between this receptor and COX-2 during inflammation [54][55]. Prostaglandins exert anti-inflammatory effects by inhibiting NF-κB mediated by the blockage of IkappaB kinase and activation of PPARγ [56].
Other evidence suggested the role of the cannabinoid system in IBD, since cannabinoid receptors 1 and 2 (CB1 and CB2) are increased in IBD colonic tissue [57]. Moreover, a strong increase of endocannabinoids (especially anandamide) was found in biopsies from patients with untreated UC [58]. The results of several studies support the hypothesis of a cross-talk between PPARγ and the cannabinoid system in reducing inflammation. Liu et al. indicated PPARγ as a molecular target for synthetic cannabinoids, demonstrating its possible use in various treatments [59]. Furthermore, the sesquiterpene β-caryophyllene (BCP) can reduce DSS-induced colitis in mice with a mechanism associated with CB2 and PPARγ, which leads to the inhibition of pro-inflammatory cytokines and NF-κB [57].
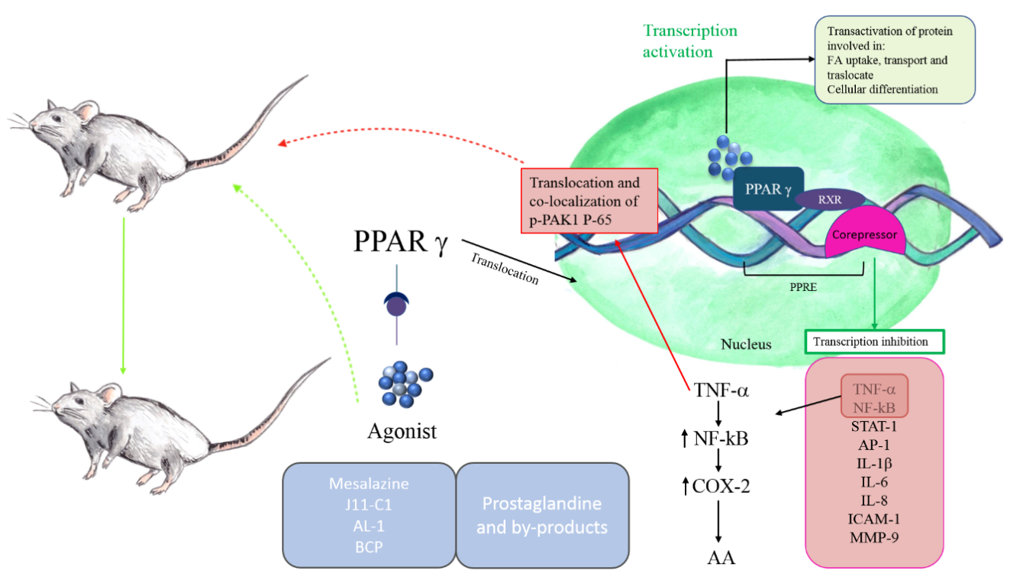
Figure 1. Peroxisome proliferator-activated receptors-γ (PPARγ) in IBDs. PPARγ belongs to the family of nuclear receptors. Its activation involves the translocation in nucleus and the heterodimerization with retinoid X receptor (RXR). The heterodimers bind to sequence-specific PPAR response elements (PPREs), stimulating the transcription of target genes. Corepressors maintain the target genes inactivated in absence of PPARγ ligands. The protective effects of PPARγ include the modulation of pro-inflammatory cytokines production, such as TNFγ, IL-6, the inhibition of transcription factors, including NF-kB, STAT-1, AP-1, and intercellular adhesion molecule and MMP-9. PPARγ also determines the downregulation of p65 expression and IkappaB kinase. In contrast, TNF-α activates NF-kB, which stimulates COX-2 to convert arachidonic acid in prostaglandins. The anti-inflammatory properties of prostaglandins are related to their ability to bind PPARγ, blocking NF-kB downstream events. PPARγ synthetic agonists can ameliorate IBD inflammation.
References
- GBD 2017 Inflammatory Bowel Disease Collaborators The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30, doi:10.1016/S2468-1253(19)30333-4.
- Torres, J.; Mehandru, S.; Colombel, J.-F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755, doi:10.1016/S0140-6736(16)31711-1.
- Gassler, N.; Rohr, C.; Schneider, A.; Kartenbeck, J.; Bach, A.; Obermüller, N.; Otto, H.F.; Autschbach, F. Inflammatory bowel disease is associated with changes of enterocytic junctions. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G216-228, doi:10.1152/ajpgi.2001.281.1.G216.
- de Souza, H.S.P.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Rev. Gastroenterol Hepatol 2016, 13, 13–27, doi:10.1038/nrgastro.2015.186.
- Neurath, M.F. Cytokines in inflammatory bowel disease. Rev. Immunol. 2014, 14, 329–342, doi:10.1038/nri3661.
- MacDermott, R.P.; Nash, G.S.; Bertovich, M.J.; Mohrman, R.F.; Kodner, I.J.; Delacroix, D.L.; Vaerman, J.-P. Altered patterns of secretion of monomeric IgA and IgA subclass 1 by intestinal mononuclear cells in inflammatory bowel disease. Gastroenterology 1986, 91, 379–385, doi:10.1016/0016-5085(86)90572-X.
- Fajas, L.; Auboeuf, D.; Raspé, E.; Schoonjans, K.; Lefebvre, A.M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.C.; Deeb, S.; et al. The organization, promoter analysis, and expression of the human PPARgamma gene. Biol. Chem. 1997, 272, 18779–18789, doi:10.1074/jbc.272.30.18779.
- Yamamoto-Furusho, J.K.; Peñaloza-Coronel, A.; Sánchez-Muñoz, F.; Barreto-Zuñiga, R.; Dominguez-Lopez, A. Peroxisome proliferator-activated receptor-gamma (PPAR-γ) expression is downregulated in patients with active ulcerative colitis: Bowel Dis. 2011, 17, 680–681, doi:10.1002/ibd.21322.
- Le Menn, G.; Neels, J.G. Regulation of Immune Cell Function by PPARs and the Connection with Metabolic and Neurodegenerative Diseases. J. Mol. Sci. 2018, 19, doi:10.3390/ijms19061575.
- Dubuquoy, L.; Rousseaux, C.; Thuru, X.; Peyrin-Biroulet, L.; Romano, O.; Chavatte, P.; Chamaillard, M.; Desreumaux, P. PPARgamma as a new therapeutic target in inflammatory bowel diseases. Gut 2006, 55, 1341–1349, doi:10.1136/gut.2006.093484.
- Huang, Q.; Chen, Q. Mediating Roles of PPARs in the Effects of Environmental Chemicals on Sex Steroids. PPAR Res. 2017, 2017, doi:10.1155/2017/3203161.
- Wahli, W.; Braissant, O.; Desvergne, B. Peroxisome proliferator activated receptors: Transcriptional regulators of adipogenesis, lipid metabolism and more. Biol. 1995, 2, 261–266, doi:10.1016/1074-5521(95)90045-4.
- Fruchart, J.-C.; Staels, B.; Duriez, P. The role of fibric acids in atherosclerosis. Atheroscler. Rep. 2001, 3, 83–92, doi:10.1007/s11883-001-0015-x.
- Ruscica, M.; Baldessin, L.; Boccia, D.; Racagni, G.; Mitro, N. Non-insulin anti-diabetic drugs: An update on pharmacological interactions. Res. 2017, 115, 14–24, doi:10.1016/j.phrs.2016.11.005.
- Berger, J.; Moller, D.E. The Mechanisms of Action of PPARs. Rev. Med. 2002, 53, 409–435, doi:10.1146/annurev.med.53.082901.104018.
- Han, L.; Shen, W.-J.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-α. Future Cardiol. 2017, 13, 259–278, doi:10.2217/fca-2016-0059.
- Moreno, S.; Farioli-Vecchioli, S.; Cerù, M.P. Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience 2004, 123, 131–145, doi:10.1016/j.neuroscience.2003.08.064.
- Sertznig, P.; Seifert, M.; Tilgen, W.; Reichrath, J. Present concepts and future outlook: Function of peroxisome proliferator-activated receptors (PPARs) for pathogenesis, progression, and therapy of cancer. Cell Physiol. 2007, 212, 1–12, doi:10.1002/jcp.20998.
- Magadum, A.; Engel, F. PPARβ/δ: Linking Metabolism to Regeneration. IJMS 2018, 19, 2013, doi:10.3390/ijms19072013.
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. J. 2014, 13, 17, doi:10.1186/1475-2891-13-17.
- Willson, T.M.; Lambert, M.H.; Kliewer, S.A. Peroxisome Proliferator–Activated Receptor γ and Metabolic Disease. Rev. Biochem. 2001, 70, 341–367, doi:10.1146/annurev.biochem.70.1.341.
- Delerive, P.; Bosscher, K.D.; Besnard, S.; Berghe, W.V.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.-C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome Proliferator-activated Receptor α Negatively Regulates the Vascular Inflammatory Gene Response by Negative Cross-talk with Transcription Factors NF-κB and AP-1. Biol. Chem. 1999, 274, 32048–32054, doi:10.1074/jbc.274.45.32048.
- Colville-Nash, P.R.; Qureshi, S.S.; Willis, D.; Willoughby, D.A. Inhibition of inducible nitric oxide synthase by peroxisome proliferator-activated receptor agonists: Correlation with induction of heme oxygenase 1. Immunol 1998, 161, 978–984.
- Chinetti, G.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors (PPARs): Nuclear receptors at the crossroads between lipid metabolism and inflammation. Res. 2000, 49, 497–505, doi:10.1007/s000110050622.
- Tan, N.S.; Michalik, L.; Noy, N.; Yasmin, R.; Pacot, C.; Heim, M.; Flühmann, B.; Desvergne, B.; Wahli, W. Critical roles of PPARβ/δ in keratinocyte response to inflammation. Genes Dev. 2001, 15, 3263–3277, doi:10.1101/gad.207501.
- Barish, G.D.; Atkins, A.R.; Downes, M.; Olson, P.; Chong, L.-W.; Nelson, M.; Zou, Y.; Hwang, H.; Kang, H.; Curtiss, L.; et al. PPARδ regulates multiple proinflammatory pathways to suppress atherosclerosis. Natl. Acad. Sci. USA 2008, 105, 4271–4276, doi:10.1073/pnas.0711875105.
- Ricote, M.; Huang, J.T.; Welch, J.S.; Glass, C.K. The peroxisome proliferator-activated receptor(PPARgamma) as a regulator of monocyte/macrophage function. Leukoc. Biol. 1999, 66, 733–739, doi:10.1002/jlb.66.5.733.
- Rosenbaum, S.E.; Greenberg, A.S. The Short- and Long-Term Effects of Tumor Necrosis Factor-α and BRL 49653 on Peroxisome Proliferator-Activated Receptor (PPAR)γ2 Gene Expression and Other Adipocyte Genes. Endocrinol. 1998, 12, 1150–1160, doi:10.1210/mend.12.8.0151.
- Okuno, A.; Tamemoto, H.; Tobe, K.; Ueki, K.; Mori, Y.; Iwamoto, K.; Umesono, K.; Akanuma, Y.; Fujiwara, T.; Horikoshi, H.; et al. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats. Clin. Investig. 1998, 101, 1354–1361, doi:10.1172/JCI1235.
- Hevener, A.L.; Olefsky, J.M.; Reichart, D.; Nguyen, M.T.A.; Bandyopadyhay, G.; Leung, H.-Y.; Watt, M.J.; Benner, C.; Febbraio, M.A.; Nguyen, A.-K.; et al. Macrophage PPARγ is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. Clin. Investig. 2007, 117, 1658–1669, doi:10.1172/JCI31561.
- Kostadinova, R.; Wahli, W.; Michalik, L. PPARs in Diseases: Control Mechanisms of Inflammation. CMC 2005, 12, 2995–3009, doi:10.2174/092986705774462905.
- Balint, B.; Nagy, L. Selective Modulators of PPAR Activity as New Therapeutic Tools in Metabolic Diseases. EMIDDT 2006, 6, 33–43, doi:10.2174/187153006776056620.
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A.; de Muynck, K.; Heymann, F.; Adarbes, V.; Defrêne, E.; et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages☆. Hepatol. 2020, 73, 757–770, doi:10.1016/j.jhep.2020.04.025.
- Ullman, T.A.; Itzkowitz, S.H. Intestinal inflammation and cancer. Gastroenterology 2011, 140, 1807–1816, doi:10.1053/j.gastro.2011.01.057.
- Lecarpentier, Y.; Claes, V.; Vallée, A.; Hébert, J.-L. Interactions between PPAR Gamma and the Canonical Wnt/Beta-Catenin Pathway in Type 2 Diabetes and Colon Cancer. PPAR Res. 2017, 2017, 5879090, doi:10.1155/2017/5879090.
- Mazzei, J.C.; Zhou, H.; Brayfield, B.P.; Hontecillas, R.; Bassaganya-Riera, J.; Schmelz, E.M. Suppression of intestinal inflammation and inflammation-driven colon cancer in mice by dietary sphingomyelin: Importance of peroxisome proliferator-activated receptor γ expression. Nutr. Biochem. 2011, 22, 1160–1171, doi:10.1016/j.jnutbio.2010.09.017.
- Dou, X.; Xiao, J.; Jin, Z.; Zheng, P. Peroxisome proliferator-activated receptor-γ is downregulated in ulcerative colitis and is involved in experimental colitis-associated neoplasia. Lett. 2015, 10, 1259–1266, doi:10.3892/ol.2015.3397.
- Gregorio, J.D.; Sferra, R.; Speca, S.; Vetuschi, A.; Dubuquoy, C.; Desreumaux, P.; Pompili, S.; Cristiano, L.; Gaudio, E.; Flati, V.; et al. Role of glycogen synthase kinase-3β and PPAR-γ on epithelial-to-mesenchymal transition in DSS-induced colorectal fibrosis. PLoS ONE 2017, 12, e0171093, doi:10.1371/journal.pone.0171093.
- McAlpine, C.A.; Barak, Y.; Matise, I.; Cormier, R.T. Intestinal-specific PPARγ deficiency enhances tumorigenesis in ApcMin/+ mice. J. Cancer 2006, 119, 2339–2346, doi:10.1002/ijc.22115.
- Stolfi, C.; Pallone, F.; Monteleone, G. Colorectal Cancer Chemoprevention by Mesalazine and Its Derivatives. Biomed. Biotechnol. 2012, 2012, doi:10.1155/2012/980458.
- Desreumaux, P.; Ghosh, S. Review article: Mode of action and delivery of 5-aminosalicylic acid—New evidence. Pharmacol. Ther. 2006, 24 (Suppl. S1), 2–9, doi:10.1111/j.1365-2036.2006.03069.x.
- Schwab, M.; Reynders, V.; Loitsch, S.; Shastri, Y.M.; Steinhilber, D.; Schröder, O.; Stein, J. PPARgamma is involved in mesalazine-mediated induction of apoptosis and inhibition of cell growth in colon cancer cells. Carcinogenesis 2008, 29, 1407–1414, doi:10.1093/carcin/bgn118.
- Sugawara, K.; Olson, T.S.; Moskaluk, C.A.; Stevens, B.K.; Hoang, S.; Kozaiwa, K.; Cominelli, F.; Ley, K.F.; McDuffie, M. Linkage to peroxisome proliferator-activated receptor-γ in SAMP1/YitFc mice and in human Crohn’s disease. Gastroenterology 2005, 128, 351–360, doi:10.1053/j.gastro.2004.11.001.
- Puleston, J.; Cooper, M.; Murch, S.; Bid, K.; Makh, S.; Ashwood, P.; Bingham, A.H.; Green, H.; Moss, P.; Dhillon, A.; et al. A distinct subset of chemokines dominates the mucosal chemokine response in inflammatory bowel disease. Pharmacol. Ther. 2005, 21, 109–120, doi:10.1111/j.1365-2036.2004.02262.x.
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82, doi:10.1038/34178.
- Vetuschi, A.; Pompili, S.; Gaudio, E.; Latella, G.; Sferra, R. PPAR-γ with its anti-inflammatory and anti-fibrotic action could be an effective therapeutic target in IBD. Rev. Med. Pharmacol. Sci. 2018, 22, 8839–8848, doi:10.26355/eurrev_201812_16652.
- Bassaganya-Riera, J.; Reynolds, K.; Martino-Catt, S.; Cui, Y.; Hennighausen, L.; Gonzalez, F.; Rohrer, J.; Benninghoff, A.U.; Hontecillas, R. Activation of PPAR γ and δ by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology 2004, 127, 777–791, doi:10.1053/j.gastro.2004.06.049.
- Choo, J.; Lee, Y.; Yan, X.; Noh, T.H.; Kim, S.J.; Son, S.; Pothoulakis, C.; Moon, H.R.; Jung, J.H.; Im, E. A Novel Peroxisome Proliferator-activated Receptor (PPAR)γ Agonist 2-Hydroxyethyl 5-chloro-4,5-didehydrojasmonate Exerts Anti-Inflammatory Effects in Colitis. Biol. Chem. 2015, 290, 25609–25619, doi:10.1074/jbc.M115.673046.
- Yang, Y.; Yan, H.; Jing, M.; Zhang, Z.; Zhang, G.; Sun, Y.; Shan, L.; Yu, P.; Wang, Y.; Xu, L. Andrographolide derivative AL-1 ameliorates TNBS-induced colitis in mice: Involvement of NF-кB and PPAR-γ signaling pathways. Rep. 2016, 6, doi:10.1038/srep29716.
- Dammann, K.; Khare, V.; Lang, M.; Claudel, T.; Harpain, F.; Granofszky, N.; Evstatiev, R.; Williams, J.M.; Pritchard, D.M.; Watson, A.; et al. PAK1 modulates a PPARγ/NF-κB cascade in intestinal inflammation. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 2349–2360, doi:10.1016/j.bbamcr.2015.05.031.
- Wang, D.; Dubois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788, doi:10.1038/onc.2009.421.
- Shattuck-Brandt, R.L.; Varilek, G.W.; Radhika, A.; Yang, F.; Washington, M.K.; DuBois, R.N. Cyclooxygenase 2 expression is increased in the stroma of colon carcinomas from IL-10(-/-) mice. Gastroenterology 2000, 118, 337–345, doi:10.1016/s0016-5085(00)70216-2.
- Singer, I.I.; Kawka, D.W.; Schloemann, S.; Tessner, T.; Riehl, T.; Stenson, W.F. Cyclooxygenase 2 is induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterology 1998, 115, 297–306, doi:10.1016/s0016-5085(98)70196-9.
- Negishi, M.; Katoh, H. Cyclopentenone prostaglandin receptors. Prostaglandins Other Lipid Mediat. 2002, 68–69, 611–617, doi:10.1016/S0090-6980(02)00059-X.
- Eibl, G. The Role of PPAR-γ and Its Interaction with COX-2 in Pancreatic Cancer. PPAR Res. 2008, 2008, doi:10.1155/2008/326915.
- Scher, J.U.; Pillinger, M.H. The anti-inflammatory effects of prostaglandins. Investig. Med. 2009, 57, 703–708, doi:10.2310/JIM.0b013e31819aaa76.
- Bento, A.F.; Marcon, R.; Dutra, R.C.; Claudino, R.F.; Cola, M.; Pereira Leite, D.F.; Calixto, J.B. β-Caryophyllene Inhibits Dextran Sulfate Sodium-Induced Colitis in Mice through CB2 Receptor Activation and PPARγ Pathway. J. Pathol. 2011, 178, 1153–1166, doi:10.1016/j.ajpath.2010.11.052.
- D’Argenio, G.; Valenti, M.; Scaglione, G.; Cosenza, V.; Sorrentini, I.; Di Marzo, V. Up-regulation of anandamide levels as an endogenous mechanism and a pharmacological strategy to limit colon inflammation. FASEB J. 2006, 20, 568–570, doi:10.1096/fj.05-4943fje.
- Liu, J.; Li, H.; Burstein, S.H.; Zurier, R.B.; Chen, J.D. Activation and binding of peroxisome proliferator-activated receptor gamma by synthetic cannabinoid ajulemic acid. Pharmacol. 2003, 63, 983–992, doi:10.1124/mol.63.5.983.

