 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Emil Bulatov | + 2049 word(s) | 2049 | 2021-02-22 07:25:52 | | | |
| 2 | Karina Chen | Meta information modification | 2049 | 2021-03-01 10:38:15 | | | | |
| 3 | Conner Chen | Meta information modification | 2049 | 2021-10-11 08:57:31 | | |
Video Upload Options
The aging of the world population leads to a constant increase of cancer-related morbidity and mortality. Treatment of late-stage tumors has become a significant burden on the healthcare system globally. Adoptive cell immunotherapy is supposed to prolong life with cancer and ideally cure cancer after a single infusion of the cell product. Arguably, the most impressive clinical therapy in this field is based on chimeric antigen receptor (CAR) T-cells capable of curing up to 25–50% of previously incurable patients with B-cell malignancies. Diverse cell therapies are already efficiently used in clinics for cancer treatment (such as tumor infiltrating lymphocytes, transgenic αβ T-cells) and several novel promising cell therapies are in development (such as CAR M-cells, transgenic γδ T-cells, CAR NK-cells). Here, we summarize the recent literature data with the focus on T-cell receptor-based therapies and overview the most advanced systems for manufacturing of clinical grade cell products.
1. Introduction
Adoptive immunotherapy is a highly potent option for the treatment of tumors resistant to the current standards of care. Chimeric antigen receptor (CAR) T-cell therapy—one of the brightest success stories in this field—has revolutionized the treatment of resistant hematological malignancies and quickly became a new standard of treatment for relapsed/refractory disease. Nevertheless, the success of CAR T-cell therapy for the treatment of solid tumors is still relatively modest. The caveats may lie in the difficulty of T-cell trafficking into solid tumor tissues due to the stromal barriers, tumor microenvironment, and certain tumor mutations resulting in activation of T-cell exclusion and exhaustion mechanisms. The second important challenge for successful application of CAR T-cell immunotherapy against solid tumors is a limited number of surface tumor-specific targets. Well-studied T-cell receptor and Natural Killer (NK) cell receptor-based technologies are highly promising in expanding the range of poten-tial tumor-specific targets. In addition, novel approaches addressing the trafficking issues were recently proposed (e.g., macrophage-derived CAR M-cells). Numerous in-stitutions and companies around the globe are working on resolving the immunother-apy accessibility issues by developing universal allogeneic cell technologies through exploitation of alternative cell sources or downregulation of cognate T-cell receptors (TCRs). The same problem is being addressed through development of automated Point-of-Care (PoC) systems for manufacturing of cell products that allow easier Good Manufacturing Practice (GMP) and biosafety compliance in comparison with the tra-ditional bioreactors.
2. The Importance of Cell Source for Production of Conventional CAR T-Cells
The manufacturing of currently approved CAR T-cell therapies results in highly heterogeneous cell products. Kymriah (by Novartis) and Yescarta (by Gilead) are produced from bulk mononuclear fraction and do not undergo any T-cell selection step. In July 2020, Tecartus (brexucabtagene autoleucel) by Gilead became the third clinically approved CAR T-cell therapy and is used for the treatment of relapsed mantle cell lymphoma. The primary difference between Yescarta and Tecartus consists in the T-cell selection step that allows depletion of circulating tumor B-cells to prevent their viral transduction and, therefore, potential tumor resistance [1]. Importantly, production of neither of the approved CAR T-cell therapies involves any further T-cell subtype selection (e.g., CD4+, CD8+, or specific T-cell memory subsets) or depletion (Treg). Thus, the precise dosage of fully functional and potent cells within the product gets specified only directly before the infusion.
The preference of several T-cell subpopulations for production of potent CAR T-cells has been previously discussed in the literature. Several studies report a variety of T-cell subpopulations, such as central memory T-cells (TCMs) [2], stem or stem-like memory T-cells (TSCMs) [3][4][5], CD27+CD45RO–CD8+ cells [4], IL17A-producing polyfunctional CD4+ T-cells [6], or defined CD4+:CD8+ (e.g., 1:1) composition [7][8] that are associated with enhanced in vitro or clinical efficacy. The unequal anti-tumor potential of certain CAR T-cell clones can be illustrated by a significant reduction of CAR T-cell clonal diversity in peripheral blood over time and eventual dominance of a small number of clones [9]. Moreover, in one particular case, the tumor was completely eliminated by a single T-cell clone that underwent tremendous expansion due to occasional disruption of the TET2 gene after lentiviral vector integration [10] without any signs of the insertional oncogenesis after 4.2 years. This report demonstrated that a single pre-selected or genetically enhanced CAR T-cell can potentially proliferate and provide a robust anti-tumor response. Therefore, safety and efficacy of the therapy to some extent relies on the careful choice of T-cell subsets for introduction of CAR and other genetic modifications [11]. Moreover, the defined CD4+:CD8+ (e.g., 1:1) composition provides additional capabilities by introduction of different CAR designs to each of the cell subtypes. The functional utility of such a complex design was proved by Guedan et al., who showed that ICOS-costimulated CD4+ CAR T-cells significantly improved the persistence of CD8+ CAR T-cells with other costimulatory domains in mice [12]. Such observations may have a significant impact on the prospective designs of clinical grade CAR T-cell products.
Within the context of solid tumors and the associated immunosuppressive microenvironment, T-cells demonstrate reduced trafficking as well as enhanced inhibition and exhaustion [13]. Given that T-cells are not the only cell subtype suitable for CAR introduction, other cell populations related to the innate immunity should be carefully considered (Figure 1).
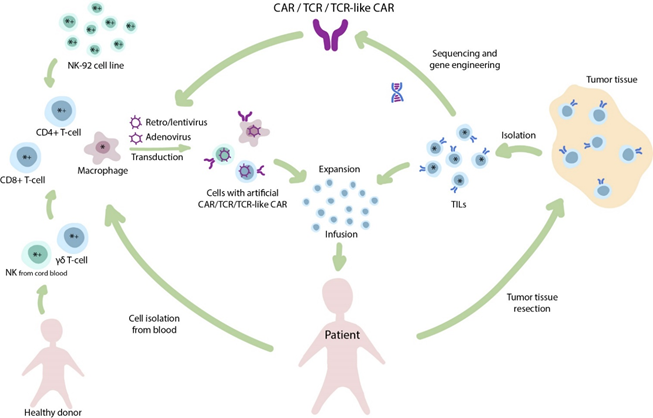
Figure 1. The diverse nature of adoptive immunotherapy. Infusion of tumor infiltrating lymphocytes (TILs) is one of the oldest clinical approaches in T-cell-based immunotherapy. The isolated TILs are expanded ex vivo and infused back to the patient. In addition, TILs and other T-cells may be used for isolation of T-cell receptors (TCRs) for further genetic engineering and generation of transgenic TCR therapies. Alternatively, to widespread CAR T-cells, other cell populations (e.g., NK-cells, macrophages) may be transduced to produce CAR NK-cells and CAR M-cells, respectively. Cellular therapies that are primarily applied for the treatment of solid tumors (*) or hematological malignancies (+).
3. CAR Cells, but Not CAR αβ T-Cells
3.1. γδ. T-Cells
The specific γδ T-cell subset attracts a growing interest in the scientific community since these cells generally recognize non-protein ligands, stress-ligands, and metabolites represented by non-conventional MHC such as CD1c or CD1d [14], as well as butyrophilin-2A1 (BTN2A1) [15] and other molecules. The British TC Biopharm and the Dutch Gadeta exploit the opportunities of this cell type for TCR and CAR-based therapies. The major advantage of these cells is stipulated by the decreased risk of autoimmune complications due to their outstanding cognate specificity (HLA-independent recognition of a narrow range of highly conservative antigens).
3.2. NK-Cells
NK-cells in contrast to T-cells recognize the presence of self-MHC class I molecules and detect stress-induced ligands on the tumor cells. However, NK-cells represent a promising cell source for CAR-based therapy because similarly to T-cells, they are capable of perforin/granzyme-dependent cytotoxicity that is regulated by activation and inhibition of surface NK-cell receptors. Interestingly, the cytokine-activated autologous peripheral blood NK-cells may possess substantial therapeutic potential even in the absence of any genetic modification (e.g., introduction of CAR), as was supported by demonstration of measurable clinical benefit in patients with relapsed glioblastoma [16][17]. The clinical benefit from application of genetically unmodified allogeneic NK-cells appears to be more prominent due to a mismatch in repertoire of killer cell immunoglobulin-like receptors (KIR) [18]. The therapeutic depletion of graft αβ T-cells prior to allogeneic hematopoietic stem cell transplantation (HSCT) is supported by the assumption that the primary graft-versus-leukemia effect originates from NK-cells and γδ T-cells rather than αβ T-cells. The follow-up of αβ and CD19+ depleted haploidentical HSCT demonstrated lower probability of chronic graft-versus-host disease (GvHD) with comparable leukemia-free survival in a multicenter retrospective trial in Italy with 343 participants [19] and some other smaller trials [20][21]. Moreover, some KIR allelic variants, such as KIR2DS4∗ 00,101 or KIR2DS2, are found to be protective in glioblastoma, while being associated with prolonged overall survival (OS) or reduced risk of glioblastoma in healthy donors [22]. Similarly, to γδ T-cells, the advantage of NK-cells as a cell source for universal allogeneic therapy is their MHC-independence and no need for knockout of endogenous TCR. On the other hand, the proliferative capacity of NK-cells is lower than that of T-cells, therefore making isolation and expansion of NK-cells to sufficient numbers a challenging task.
In a clinical setting, the increased attention to allogeneic NK-cells, e.g., based on the NK-92 line initially derived from a lymphoma patient, is explained by a limited proliferative capacity of primary donor-derived NK-cells. Obviously, such allogeneic cells require prior irradiation to prevent their inoculation leading to formation of secondary tumors. Preclinical [23] trials demonstrated the increased in vitro and in vivo activity of HER2-specific CAR-NK-92 cells in comparison to the parental NK-92. Further, a number of clinical trials is ongoing and a case report of successful treatment of relapsed glioblastoma patients with surgery and HER2-specific CAR-NK-92 cells was presented by Dr. W. Wels to illustrate phase I CAR2BRAIN (NCT03383978) clinical trial [24]. The therapeutic application of CD33-CAR-NK-92 cells in Phase I trial with dose up to 5 × 109 cells was shown to be safe although the clinical benefit was only moderate (significant but transient reduction in blast count) suggesting its most appropriate application as a “bridge” to allogeneic HSCT [25].
Exploiting cord blood allogeneic donor-derived NK-cells allowed Liu et al. at MD Anderson Cancer Center to demonstrate the preliminary efficacy of CAR NK-cells to be almost equivalent to CAR T-cells, at least for B-cell malignancies and in a short period of time [26]. CAR-transduced cord blood NK-cells are designed to produce IL-15 for self-stimulation and, in contrast to NK-92, lack tumorigenic potential that allows omission of the irradiation step during preparation of the therapeutic cell product. The results of the Phase I/II trial revealed 8 out of 11 responses and 7 complete remissions, while some patients demonstrated persistence of the minimal residual disease (MRD)—a small number of tumor cells in the blood, as confirmed by flow cytometry. The complexity of clinical implementation might explain the fact that to date, there are only six ongoing clinical trials with NK-92 cells and only two trials with cord blood NK cells.
3.3. CAR M-Cells
Low penetration capability of CAR T-cells into the solid tumor tissues is well known and was extensively discussed in the literature [13][27][28][29]. Adusumilli et al. report observations from a preclinical model demonstrating that intrapleurally administration of CAR T-cells specific to mesothelin required up to 30-fold fewer cells to induce long-term complete remission compared to systemically infused CAR T-cells [30]. This issue of CAR T-cell therapy was addressed in an immunotherapy approach developed specifically for the treatment of solid tumors whereby an adenoviral first-generation CAR vector is transduced in human macrophages resulting in CAR M-cells [31]. These macrophage-derived cells were capable of efficient trafficking to the tumor site, target-specific phagocytosis and reprogramming of the tumor microenvironment (TME) while retaining M1 self-polarization. Importantly, it was found that CAR M-cells are able to induce epitope spreading, the expansion mechanism of tumor-specific T-cells that functions through enhanced processing and presentation of tumor epitopes as a result of phagocytosis by CAR M-cells. Overall, CAR-transduced macrophages significantly improved the survival rates of immunodeficient NOD/SCID mice. Moreover, they have a potential for even stronger impact on the survival of immunocompetent mice with functional T-cells due to the epitope spreading effect [32].
With regards to potential clinical application, macrophages are known to be a hardly susceptible for lentiviral transduction due to the low proliferations rates [33][34]. In ovine models, the efficient transduction was achieved only at very high multiplicity of infection (MOI) values of 10–60 and with the use of polycations like hexadimethrine bromide (polybrene) or protamine sulfate [35]. For CAR M-cells, Klichinsky et al. demonstrated high transduction efficiency using adenoviral vector (serotype 5) [31]. In principle, adenoviral vectors appear to be safe and are used in clinic for oncolytic viral therapy[36] and as vaccines, including anti-SARS CoV-2 [37][38]. However, inability of adenovirus to integrate the cell genome might result in low persistence of CAR expression in adenovirus-transduced macrophages. In addition to that, such critical points as the number of cells per infusion and the potential toxicity remain to be more thoroughly investigated in future clinical trials.
To summarize, further development of the advanced ACT approaches mentioned above is particularly important in the context of two primary issues associated with CAR T-cell therapy: (i) limited affordability for patients due to the high costs of personalized production and the lack of universal allogeneic clinical-stage therapies; (ii) low efficacy against solid tumors (e.g., glioblastoma or pancreatic cancer) that can be potentially resolved by using CAR M-cells associated with enhanced tumor penetration and positive impact on endogenous T-cell response.
References
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kuli-kovskaya, I.; et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018, 24, 1499–1503, doi:10.1038/s41591-018-0201-9.
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Anti-tumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056, doi:10.1182/blood-2011-05-354449.
- Schmueck-Henneresse, M.; Omer, B.; Shum, T.; Tashiro, H.; Mamonkin, M.; Lapteva, N.; Sharma, S.; Rollins, L.; Dotti, G.; Reinke, P.; et al. Comprehensive Approach for Identifying the T Cell Subset Origin of CD3 and CD28 Antibody–Activated Chimeric Antigen Receptor–Modified T Cells. J. Immunol. 2017, 199, 348–362, doi:10.4049/jimmunol.1601494.
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571, doi:10.1038/s41591-018-0010-1.
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259, doi:10.1038/s41591-019-0522-3.
- Rossi, J.; Paczkowski, P.; Shen, Y.-W.; Morse, K.; Flynn, B.; Kaiser, A.; Ng, C.; Gallatin, K.; Cain, T.; Fan, R.; et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 2018, 132, 804–814, doi:10.1182/blood-2018-01-828343.
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell all patients. J. Clin. Invest. 2016, 126, 2123–2138, doi:10.1172/JCI85309.
- Abramson, J.S.; Lia, P.M.; Gordon, L.I.; Lunning, M.A.; Arnason, J.E.; Wang, M. High Durable CR Rates in Re-lapsed/Refractory (R/R) Aggressive B-NHL Treated with the CD19-Directed CAR T Cell Product JCAR017 (TRANSCEND NHL 001): Defined Composition Allows for Dose-Finding and Definition of Pivotal Cohort. In Proceedings of the 2017 An-nual American Society for Hematology Meeting, Atlanta, GA, USA, 9–12 December 2017.
- Sheih, A.; Voillet, V.; Hanafi, L.A.; DeBerg, H.A.; Yajima, M.; Hawkins, R.; Gersuk, V.; Riddell, S.R.; Maloney, D.G.; Wohlfahrt, M.E.; et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun. 2020, 11, 1–13, doi:10.1038/s41467-019-13880-1.
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312, doi:10.1038/s41586-018-0178-z.
- Titov, A.; Petukhov, A.; Staliarova, A.; Motorin, D.; Bulatov, E.; Shuvalov, O.; Soond, S.M.; Piacentini, M.; Melino, G.; Za-ritskey, A.; et al. The biological basis and clinical symptoms of CAR-T therapy-associated toxicites. Cell Death Dis. 2018, 9, 897, doi:10.1038/s41419-018-0918-x.
- Guedan, S.; Posey, A.D.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, doi:10.1172/jci.insight.96976.
- Titov, A.; Valiullina, A.; Zmievskaya, E.; Zaikova, E.; Petukhov, A.; Miftakhova, R.; Bulatov, E.; Rizvanov, A. Advancing CAR T-cell therapy for solid tumors: Lessons learned from lymphoma treatment. Cancers 2020, 12, doi:10.3390/cancers12010125.
- Barral, D.C.; Brenner, M.B. CD1 antigen presentation: How it works. Nat. Rev. Immunol. 2007, 7, 929–941.
- Karunakaran, M.M.; Willcox, C.R.; Salim, M.; Paletta, D.; Fichtner, A.S.; Noll, A.; Starick, L.; Nöhren, A.; Begley, C.R.; Ber-wick, K.A.; et al. Butyrophilin-2A1 Directly Binds Germline-Encoded Regions of the Vγ9Vδ2 TCR and Is Essential for Phos-phoantigen Sensing. Immunity 2020, 52, 487-498.e6, doi:10.1016/j.immuni.2020.02.014.
- Ishikawa, E.; Tsuboi, K.; Saijo, K.; Harada, H.; Takano, S.; Nose, T.; Ohno, T. Autologous natural killer cell therapy for hu-man recurrent malignant glioma. Anticancer Res. 2004, 24, 1861–1871.
- Yoshida, S.; Tanaka, R.; Takai, N.; Ono, K. Local Administration of Autologous Lymphokine-activated Killer Cells and Re-combinant Interleukin 2 to Patients with Malignant Brain Tumors. Cancer Res. 1988, 48.
- Lupo, K.B.; Matosevic, S. Natural killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers 2019, 11, doi:10.3390/cancers11060769.
- Bertaina, A.; Zecca, M.; Buldini, B.; Sacchi, N.; Algeri, M.; Saglio, F.; Perotti, C.; Gallina, A.M.; Bertaina, V.; Lanino, E.; et al. Unrelated donor vs. HLA-haploidentical a/b T-cell– and B-cell–depleted HSCT in children with acute leukemia. Blood 2018, 132, 2594–2607, doi:10.1182/blood-2018-07-861575.
- Shah, R.M.; Elfeky, R.; Nademi, Z.; Qasim, W.; Amrolia, P.; Chiesa, R.; Rao, K.; Lucchini, G.; Silva, J.M.F.; Worth, A.; et al. T-cell receptor αβ+ and CD19+ cell–depleted haploidentical and mismatched hematopoietic stem cell transplantation in pri-mary immune deficiency. J. Allergy Clin. Immunol. 2018, 141, 1417–1426.e1, doi:10.1016/j.jaci.2017.07.008.
- Dovydenko, M.V.; Parovichnikova, E.N.; Kuzmina, L.A.; Vasilyeva, V.A.; Drokov, M.Y.; Koroleva, O.M.; Mikhaltsova, E.D.; Popova, N.N.; Konova, Z.V.; Dmitrova, A.A.; et al. Haploidentical Stem Cell Transplantation with TCR Alpha/Beta and CD19 Depletion in Adult Patients with Hematological Malignancies. Blood 2019, 134, 5648–5648, doi:10.1182/blood-2019-131316.
- Burger, M.C.; Zhang, C.; Harter, P.N.; Romanski, A.; Strassheimer, F.; Senft, C.; Tonn, T.; Steinbach, J.P.; Wels, W.S. CAR-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors into Precision Tools for Cancer Im-munotherapy. Front. Immunol. 2019, 10, 2683.
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108, doi:10.1093/jnci/djv375.
- Wels W. (2020, January), CAR-NK cells as off-the-shelf therapeutics bridging innate and adaptive immunity, Presented at 2nd European CAR T-cell meeting, Barcelona, Spain.
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-Man Clinical Trial of CAR NK-92 Cells: Safety Test of CD33-CAR NK-92 Cells in Patients with Relapsed and Refractory Acute Myeloid Leukemia; e-Century Publishing Corporation: Madison, WI, USA, 2018; Volume 8.
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553, doi:10.1056/NEJMoa1910607.
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167, doi:10.1038/s41571-018-0142-8.
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase pro-motes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529, doi:10.1038/nm.3833.
- Springuel, L.; Lonez, C.; Alexandre, B.; Van Cutsem, E.; Machiels, J.P.H.; Van Den Eynde, M.; Prenen, H.; Hendlisz, A.; Shaza, L.; Carrasco, J.; et al. Chimeric Antigen Receptor-T Cells for Targeting Solid Tumors: Current Challenges and Existing Strategies. BioDrugs 2019, 33, 515–537, doi:10.1007/s40259-019-00368-z.
- Adusumilli, P.S.; Cherkassky, L.; Villena-Vargas, J.; Colovos, C.; Servais, E.; Plotkin, J.; Jones, D.R.; Sadelain, M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 2014, 6, doi:10.1126/scitranslmed.3010162.
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, doi:10.1038/s41587-020-0462-y.
- Chulpanova, D.S.; Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Mouse Tumor Models for Advanced Can-cer Immunotherapy. Int. J. Mol. Sci. 2020, 21, 4118, doi:10.3390/ijms21114118.
- Zhang, X.; Edwards, J.P.; Mosser, D.M. The expression of exogenous genes in macrophages: Obstacles and opportunities. Methods Mol. Biol. 2009, 531, 123–143, doi:10.1007/978-1-59745-396-7_9.
- Bobadilla, S.; Sunseri, N.; Landau, N.R. Efficient transduction of myeloid cells by an HIV-1-derived lentiviral vector that packages the Vpx accessory protein. Gene Ther. 2013, 20, 514–520, doi:10.1038/gt.2012.61.
- Karponi, G.; Kritas, S.; Petridou, E.; Papanikolaou, E. Efficient transduction and expansion of ovine macrophages for gene therapy implementations. Vet. Sci. 2018, 5, doi:10.3390/vetsci5020057.
- Hajeri, P.B.; Sharma, N.S.; Yamamoto, M. Oncolytic Adenoviruses: Strategies for Improved Targeting and Specificity. Cancers 2020, 12, 1504, doi:10.3390/cancers12061504.
- Zhu, F.C.; Guan, X.H.; Li, Y.H.; Huang, J.Y.; Jiang, T.; Hou, L.H.; Li, J.X.; Yang, B.F.; Wang, L.; Wang, W.J.; et al. Immuno-genicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2020, 396, 479–488, doi:10.1016/S0140-6736(20)31605-6.
- Logunov, D.Y.; Dolzhikova, I.V.; Zubkova, O.V.; Tukhvatullin, A.I.; Shcheblyakov, D.V.; Dzharullaeva, A.S.; Grousova, D.M.; Erokhova, A.S.; Kovyrshina, A.V.; Botikov, A.G.; et al. Safety and immunogenicity of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine in two formulations: Two open, non-randomised phase 1/2 studies from Russia. Lancet 2020, 396, 887–897, doi:10.1016/S0140-6736(20)31866-3.

