 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Elisabeth Taucher | + 2310 word(s) | 2310 | 2021-02-19 03:06:48 | | | |
| 2 | Bruce Ren | -21 word(s) | 2289 | 2021-02-23 02:48:08 | | |
Video Upload Options
Non-small cell lung cancer (NSCLC) accounts for the majority of lung cancer subtypes. Two to seven percent of NSCLC patients harbor gene rearrangements of the anaplastic lymphoma kinase (ALK) gene or, alternatively, harbor chromosomal fusions of ALK with echinoderm microtu-bule-associated protein-like 4 (EML4). The availability of tyrosine kinase inhibitors targeting ALK (ALK-TKIs) has significantly improved the progression-free and overall survival of NSCLC pa-tients carrying the respective genetic aberrations.
1. Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide [1]. Approximately 80–85% of lung cancers count among non-small cell histology (NSCLC) [1], and approximately 2–7% of NSCLC cases feature positivity for anaplastic lymphoma kinase (ALK) gene rearrangement or connection with echinoderm microtubule-associated protein-like 4 (EML4) [2][3]. Fusion with the EML4 gene remains the most common form of ALK alteration [4]. NSCLC patients featuring ALK-EML4 gene fusion are very sensitive to treatment with ALK tyrosine kinase inhibitors (ALK-TKIs). Fusion of the ALK-EML4 genes in NSCLC can be detected in tumor samples by means of various methods, first of all fluorescence in situ hybridization (FISH), or else quantitative reverse transcriptase polymerase chain reaction (qRT-PCT) and immunohistochemistry (IHC) [5]. The disadvantage of RT-PCR is that it highly depends on RNA quality, which is often less than ideal in formalin-fixed and paraffin-embedded tissue samples [6][7]. However, next-generation sequencing (NGS) panels, such as the Archer®FusionPlex® panel provide an effective alternative method for the detection of both known and novel ALK gene rearrangements with great accuracy [8]. In most specialized centers, ALK gene rearrangement analysis has become standard in the diagnostic workup of NSCLC, and ALK-inhibitors are increasingly used for NSCLC treatment in this particular subtype. According to a meta-analysis by Li et al., ALK-inhibitors were found to significantly improve the overall survival (OS) and progression free survival (PFS) in NSCLC patients, especially in patients whose tumors harbor ALK- or ROS1 gene fusions [5]. Median OS for ALK-positive NSCLC patients has nowadays increased to seven years [9][10], being currently the best reported OS of all forms of metastatic NSCLC defined by genomic variants. ALK inhibitors contributed to a better prognosis of patients, having improved one-year or two-year OS, PFS and objective response rate (ORR). Still, it has to be pointed out that ALK-positive NSCLC is a considerably aggressive subtype, mainly because of its inevitable tendency to cause brain involvement. Figure 1 shows how the EML4-ALK fusion gene is constructed (Figure 1).
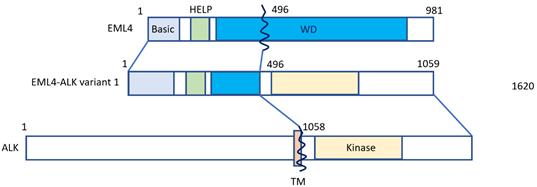
Figure 1. The EML4-ALK fusion gene. The N-terminal portion of EML4 is fused, containing the main region of EML4-ALK, i.e., the echinoderm microtubule-associated protein-like protein (HELP) domain, and part of the WD-repeat region to the intracellular region of ALK, which contains the tyrosine kinase domain. The transmembrane (TM) domain is not part of the final fusion product. Reproduced from Golding et al. [11].
That is to say, existing ALK inhibitors bind with varying contact sites in the ATP binding pocket, which results in unique and specific ways of inhibition of different ALK mutations. This was confirmed in a study on ALK mutations in neuroblastoma patients, in ALK-TKI resistant NSCLC patients and subjects suffering from myofibroblastic tumors [12]. Furthermore, the inhibition profile of different ALK-TKIs is affected by different fusion variants of EML4-ALK, or by the properties of other fusion partners [13]. In a study from Lin and colleagues from 2018, ALK variants were identified in a cohort of 129 patients, and possible links to ALK-TKI resistance were drawn [13]. EML4-ALK variant 1 was found to be the most frequent ALK variant, occurring in 43% of the investigated subjects, alongside EML4-ALK variant 3, which occurred in 40% of patients. ALK resistance mutations were much more common in variant 3, as compared to variant 1 (p = 0.023). In patients who received the third-generation ALK inhibitor lorlatinib, the EML4-ALK variant 3 was linked to a strikingly better PFS [14].
Increasing evidence suggests that NSCLC cells consequently develop resistance mechanisms against ALK-inhibitors in almost all cases, which makes it mandatory to follow up patients during the course of the disease by repeated molecular testing, especially in the case of tumor progression upon ALK-inhibitor treatment.
In Figure 2, the complex manner of interaction of the EML4-ALK protein complex is illustrated, realized using a tandem affinity purification approach followed by mass spectrometry [11] (Figure 2).
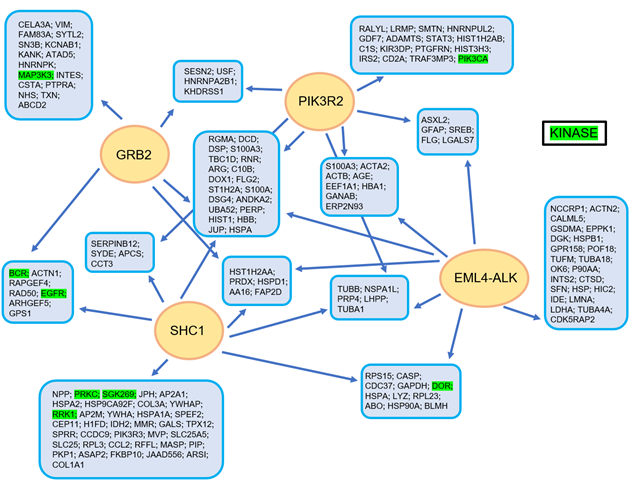
Figure 2. The EML4-ALK protein complex interaction model, as constructed using a tandem affinity purification approach with consecutive mass spectrometry. Reproduced from Golding et al. [11].
To date, more than 6,000 X-ray crystal structures have been discovered that are in the public domain of protein kinases [15]. An even larger number of three-dimensional proprietary structures are used by pharmaceutical companies for the discovery of new protein kinase inhibitors. Currently, about 175 protein kinase inhibitors that can be administered orally are being tested in clinical settings worldwide [16]. Close to 50 drugs that are directed against about 20 different protein kinases have already been approved by the Food and Drug Administration (FDA), having their points of action in about 20 different protein kinases [16][17]. Malignant cells are generally genomically unstable, and thus, resistance to protein kinase-targeting drugs occurs regularly over the disease course. As of today, it is not clear whether acquired resistance also occurs in protein kinase inhibitors when prescribed for inflammatory or autoimmune disorders [15]. All the different ALK fusion proteins feature a complex and multi-layered network of interaction with other proteins through a multitude of downstream pathways, like JAK/STAT, PI3K/AKT, or MEK/ERK [18][19]. When protein kinase inhibitors are administered over a longer time period, these complex models of interaction change in structure, leading to a dysregulation and, ultimately, acquired drug resistance [20]
2. Acquired ALK Resistance Mutations
Crizotinib, a first-generation ALK-TKI, was the first agent to be approved for clinical use. Crizotinib showed striking clinical efficacy when used as a therapeutic option in ALK-rearranged NSCLC. Recent follow-up data of clinical trials showed a response rate of >60% and a PFS of >12 months upon crizotinib therapy [21][22][23]. It has been clearly demonstrated for this agent that in nearly all patients showing good clinical response to treatment in the first place, resistance to the drug is acquired over time. Most often, secondary crizotinib resistance is due to acquired ALK gene mutations. Of note, de novo ALK resistance mutations, as well as pre-existing genetic aberrations leading to ALK-TKI therapy failure are generally rare (<3–5% of ALK-resistant NSCLC) [24]. Unlike epidermal growth factor (EGFR)-TKI resistance in NSCLC harboring EGFR mutations, where one single (EGFR T790M mutation) is outlined in about 60% of patients resistant to treatment, various ALK-resistance mutations (e.g., L1196M, I1171T/N/S, L1152P/R, F1174C/L/V, C1156Y/T, I1171T/N/S, S1206C/Y, G1269A/S, V1180L and 19 G1202R) are found in 20–25% of treatment-resistant subjects [25]. Second-generation ALK-TKIs, i.e., alectinib [26][27] and ceritinib [28], characterized by a different sensitivity-spectrum to ALK resistance mutations, have been approved as a treatment of NSCLC following resistance to crizotinib [25]. Various mechanisms of ALK-TKI resistance have been demonstrated so far, namely ALK gene amplification [29][30][41][42], activation of ALK via bypass signaling pathways [31][32], adopting different driver oncogenes like EGFR and BRAF [33], or insufficient drug penetration across the blood brain barrier and an enhanced expression of P-glycoprotein [34]. When these resistance mechanisms occur, other therapeutic options have to be implemented. Previous experience with crizotinib in clinical practice shows that nearly all ALK-positive patients are diagnosed with cerebral metastases sooner or later [35]. This is due to the lack of penetration of crizotinib to the blood brain barrier, even if patients still respond systemically to treatment. Moreover, only a small number of patients who develop brain metastases upon treatment with crizotinib develop ALK resistance mutations. Hence, the occurrence of brain metastases under crizotinib treatment strongly necessitated the development of second-generation ALK inhibitors.
From a molecular standpoint, the more bulky and charged side chain of the ALK kinase is assumed to cause steric interference of most ALK inhibitors [36][37][38]. ALK F1174 mutations, for instance, are located very close to the C-terminus of the alpha C helix, most likely stabilizing and activating a conformation increasing the likelihood of ALK to bind to ATP [39][40].
In a study by Gainor et al., it was investigated how frequently ALK resistance mutations occur in a cohort of 51 patients with ALK-positive tumors, who had progressive disease upon treatment with crizotinib [41]. Tissue biopsies for this analysis were mostly acquired when patients still received crizotinib, or within one month after the stop of crizotinib therapy. In only 11 (20%) of biopsy samples, ALK resistance mutations were outlined. The most common ALK resistance mutations were L1196M and G1269A, but these were present only in 7% and 4% of the samples with crizotinib restistance, respectively [41]. Other mutations identified were C1156Y (2%), G1202R (2%), I1171T (2%), S1206Y (2%), and E1210K (2%). An interesting finding from the same study was that following treatment of second-generation ALK inhibitors, resistance mutations occurred more frequently [41]. Patients with disease progression upon treatment with ceritinib (n = 23), alectinib (n = 17), or brigatinib (n = 6) were investigated regarding ALK resistance mutations. Among 23 patients with ceritinib resistance, 21 (91%) had primarily received crizotinib. In nine patients, biopsies prior to initiation of ceritinib, or after crizotinib were also available, and only two of them showed on-target mechanisms of resistance. Overall, 54% of ceritinib-resistant tumor specimens harbored ALK resistance mutations, and 17% contained more than two different ALK resistance mutations, with G1202R (21%) and F1174C/L (16.7%) being the most common ones [41]. In the same study, the authors were also able to outline ALK C1156Y mutations in two (8%) of specimens, which lessens the response to ceritinib, as previously shown. Moreover, a previously unknown ALK mutation, namely G1202del, was outlined in two tumor specimens (8%) [41]. Consecutively, in this study, Ba/F3 cells stably expressing EML4-ALK harboring the G1202del were engineered and treated with diverse ALK inhibitors. It was demonstrated that G1202del was associated with resistance to ceritinib, alectinib and brigatinib, while the efficacy of crizotinib was less affected by this deletion.
In addition, a cohort of 17 ALK-positive patients was analyzed, and all patients underwent repeated biopsies after they had developed progression upon treatment with alectinib. All 17 subjects had received crizotinib prior to alectinib [41]. In nine (53%) biopsy samples, ALK resistance mutations were outlined, the most common being G1202R, which occurred in 29% of cases. Interestingly, preclinical models have suggested alectinib to exert a considerable activity against L1196M, a known ALK-gatekeeper mutation [42], yet in this cohort of post-alectinib biopsies this mutation was observed in one subject as well. Another patient cohort of six ALK-positive subjects who had developed resistance to brigatinib was analyzed as well. ALK-resistance mutations were observed in five out of seven patients (71%). Similar to patients progressing upon ceritinib or alectinib treatment, the most common ALK resistance mutation was G1202R, which was outlined in three specimens [41]. Summing up their investigation, the authors pointed out that most patients received systemic chemotherapy (25%) or underwent enrolment into clinical trials (31%) after having developed resistance to ceritinib, alectinib and/or brigatinib. Of note, no consecutive therapy of any kind was administered in 38% of patients after they had progressed, or else no follow-up visit was done. According to a case report of a patient who relapsed upon brigatinib, the ALK-E1210K + D1203N double mutation was found [41]. However, it must be kept in mind that this patient had been treated with first-line crizotinib. Hence, no conclusions can be drawn between the findings in post-progression biopsies and the clinical response to sequential treatments. Across all three patient cohorts analyzed, 56% of ALK-positive patients progressed under treatment with second-generation ALK inhibitors (ceritinib: 54%; alectinib: 53%; brigatinib: 71%). Hence, ALK resistance mutations occurred at a significantly higher rate following therapy with the more potent second-generation ALK inhibitors when compared to a prevalence of ALK resistance mutations of only 20% in subjects who progressed under crizotinib [41].
Doebele and colleagues carried out a study aiming to outline resistance mechanisms to treatment with crizotinib. Tissue from 14 ALK-positive NSCLC patients was obtained, all of whom progressed upon crizotinib therapy, as confirmed radiologically. Molecular analysis was performed with 11 tumor specimens. Four patients (36%) developed secondary ALK tyrosine kinase domain mutations. In two of these subjects, a new ALK mutation was outlined, encoding a G1269A amino acid substitution, which had previously been linked to crizotinib resistance in vitro. A newly occurring ALK copy number gain was seen in two patients, one of whom had an ALK resistance mutation. One patient showed excessive growth of an EGFR mutant NSCLC, but an ALK gene rearrangement was not observed. Two patients featured mutation of KRAS, and one of these two patients did not have an ALK gene rearrangement. Interestingly, in one patient an ALK gene fusion negative tumor had newly developed, contrary to the baseline tumor sample. However, there was no identifiable alternative driver. Two patients retained ALK positivity in the absence of an identifiable resistance mutation. Summing up these data about crizotinib resistance in ALK-positive NSCLC, crizotinib resistance is promoted by somatic mutations of the ALK kinase domain, by ALK gene fusion copy number gain and by newly emerging individual oncogenic mutations.
Of note, according to a recent case report, a novel ROS1-FBXL17 (F-box and leucine-rich repeat protein 17) fusion, co-existing with CD74-ROS1 fusion was detected in a patient with lung adenocarcinoma, possibly improving sensitivity to crizotinib. The authors point out that today only approximately 24 ROS1 fusion partners are known that exhibit crizotinib sensitivity. However, this non-reciprocal/reciprocal ROS1 translocation, containing a novel ROS1-FBXL17 fusion co-existing with the CD74-ROS1 fusion, was found in a Chinese patient suffering from lung adenocarcinoma, who responded well to treatment with crizotinib. Interestingly, PFS of this patient was 15.7 months, exceeding the highest PFS level reported among the Chinese population so far (14.2 months). The authors of this report suggest this particular ROS1-FBXL17 fusion to synergistically promote the sensitivity of the CD74-ROS1 fusion to crizotinib. This interesting case shows that crizotinib serves as a potential treatment option for patients with double ROS1 fusions, or non-reciprocal/reciprocal ROS1 translocation.
References
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2014, 64, 9–29.
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global Survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203.
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.-I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nat. Cell Biol. 2007, 448, 561–566.
- Marsilje, T.H.; Pei, W.; Chen, B.; Lu, W.; Uno, T.; Jin, Y.; Jiang, T.; Kim, S.; Li, N.; Warmuth, M.; et al. Synthesis, Structure–Activity Relationships, and in Vivo Efficacy of the Novel Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor 5-Chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) Currently in Phase 1 and Phase 2 Clinical Trials. J. Med. Chem. 2013, 56, 5675–5690.
- Li, G.; Dai, W.-R.; Shao, F.-C. Effect of ALK-inhibitors in the treatment of non-small cell lung cancer: A systematic review and meta-analysis. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3496–3503.
- Wong, D.; Yip, S.; Sorensen, P.H. Methods for Identifying Patients with Tropomyosin Receptor Kinase (TRK) Fusion Cancer. Pathol. Oncol. Res. 2020, 26, 1385–1399.
- Teixido, C.; Giménez-Capitán, A.; Molina-Vila, M.A.; Peg, V.; Karachaliou, N.; Rodríguez-Capote, A.; Castellvi, J.; Rosell, R. RNA Analysis as a Tool to Determine Clinically Relevant Gene Fusions and Splice Variants. Arch. Pathol. Lab. Med. 2018, 142, 474–479.
- Vendrell, J.A.; Taviaux, S.; Béganton, B.; Godreuil, S.; Audran, P.; Grand, D.; Clermont, E.; Serre, I.; Szablewski, V.; Coopman, P.; et al. Detection of known and novel ALK fusion transcripts in lung cancer patients using next-generation sequencing ap-proaches. Sci. Rep. 2017, 7, 12510.
- Duruisseaux, M.; Besse, B.; Cadranel, J.; Pérol, M.; Mennecier, B.; Bigay-Game, L.; Descourt, R.; Dansin, E.; Audigier-Valette, C.; Moreau, L.; et al. Overall survival with crizotinib and next-generation ALK inhibitors in ALK-positive non-small-cell lung cancer (IFCT-1302 CLINALK): A French nationwide cohort retrospective study. Oncotarget 2017, 8, 21903–21917.
- Pacheco, J.M.; Gao, D.; Smith, D.; Purcell, T.; Hancock, M.; Bunn, P.; Robin, T.; Liu, A.; Karam, S.; Gaspar, L.; et al. Natural History and Factors Associated with Overall Survival in Stage IV ALK-Rearranged Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2019, 14, 691–700.
- Golding, B.; Luu, A.; Jones, R.; Viloria-Petit, A.M. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol. Cancer 2018, 17, 1–15.
- Umapathy, G.; Mendoza‐Garcia, P.; Hallberg, B.; Palmer, R.H. Targeting anaplastic lymphoma kinase in neuroblastoma. APMIS 2019, 127, 288–302.
- Li, X.; Li, J.; Wu, P.; Zhou, L.; Lu, B.; Ying, K.; Chen, E.; Lu, Y.; Liu, P. Smoker and non-smoker lung adenocarcinoma is characterized by distinct tumor immune microenvironments. OncoImmunology 2018, 7, e1494677.
- Lin, J.J.; Zhu, V.W.; Yoda, S.; Yeap, B.Y.; Schrock, A.B.; Dagogo-Jack, I.; Jessop, N.A.; Jiang, G.Y.; Le, L.P.; Gowen, K.; et al. Impact of EML4-ALK Variant on Resistance Mechanisms and Clinical Outcomes in ALK-Positive Lung Cancer. J. Clin. Oncol. 2018, 36, 1199–1206.
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50.
- Carles, F.; Bourg, S.; Meyer, C.; Bonnet, P. PKIDB: A Curated, Annotated and Updated Database of Protein Kinase Inhibitors in Clinical Trials. Molecules 2018, 23, 908, doi:10.3390/molecules23040908.
- Fischer, P.M. Approved and Experimental Small-Molecule Oncology Kinase Inhibitor Drugs: A Mid-2016 Overview. Med. Res. Rev. 2017, 37, 314–367.
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23.
- Zhang, G.; Scarborough, H.; Kim, J.; Rozhok, A.I.; Chen, Y.A.; Zhang, X.; Song, L.; Bai, Y.; Fang, B.; Liu, R.Z.; et al. Coupling an EML4-ALK-centric interactome with RNA interference identifies sensitizers to ALK inhibitors. Sci. Signal. 2016, 9, rs12.
- Lin, J.J.; Riely, G.J.; Shaw, A.T. Targeting ALK: Precision Medicine Takes on Drug Resistance. Cancer Discov. 2017, 7, 137–155.
- Kwak, E.L.; Bang, Y.-J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.-H.I.; Dezube, B.J.; Jänne, P.A.; Costa, D.B.; et al. Anaplastic Lymphoma Kinase Inhibition in Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2010, 363, 1693–1703.
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus Chemotherapy in AdvancedALK-Positive Lung Cancer. N. Engl. J. Med. 2013, 368, 2385–2394.
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177.
- Lucena-Araujo, A.R.; Moran, J.P.; VanderLaan, P.A.; Dias-Santagata, R.; Folch, E.; Majid, A.; Kent, M.S.; Gangadharan, S.; Rangachari, D.; Huberman, M.S.; et al. De novo ALK kinase domain mutations are uncommon in kinase inhibitor-naïve ALK rearranged lung cancers. Lung Cancer 2016, 99, 17–22.
- Fukuda, K.; Takeuchi, S.; Arai, S.; Katayama, R.; Nanjo, S.; Tanimoto, A.; Nishiyama, A.; Nakagawa, T.; Taniguchi, H.; Suzuki, T.; et al. Epithelial-to-Mesenchymal Transition Is a Mechanism of ALK Inhibitor Resistance in Lung Cancer Independent of ALK Mutation Status. Cancer Res. 2019, 79, 1658–1670.
- Gadgeel, S.M.; Gandhi, L.; Riely, G.J.; Chiappori, A.A.; West, H.L.; Azada, M.C.; Morcos, P.N.; Lee, Ru.; Garcia, L.; Yu, L.; et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): Results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014, 15, 1119–1128.
- Seto, T.; Kiura, K.; Nishio, M.; Nakagawa, K.; Maemondo, M.; Inoue, A.; Hida, T.; Yamamoto, N.; Yoshioka, H.; Harada, M.; et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): A single-arm, open-label, phase 1–2 study. Lancet Oncol. 2013, 14, 590–598.
- Shaw, A.T.; Kim, T.M.; Crinò, L.; Gridelli, C.; Kiura, K.; Liu, G.; Novello, S.; Bearz, A.; Gautschi, O.; Mok, T.; et al. Ceritinib versus chemotherapy in patients with ALK-rearranged non-small-cell lung cancer previously given chemotherapy and crizo-tinib (ASCEND-5): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 874–886.
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med. 2012, 4, 120ra17.
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular mechanisms of resistance to first- and second-generation alk inhibitors in alk-rearranged lung cancer. Cancer Discov. 2016, 6, 1118–1133.
- Lovly, C.M.; McDonald, N.T.; Chen, H.; Ortiz-Cuaran, S.; Heukamp, L.C.; Yan, Y.; Florin, A.; Ozretić, L.; Lim, D.; Wang, L.; et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion–positive lung cancer. Nat. Med. 2014, 20, 1027–1034.
- Yamada, T.; Takeuchi, S.; Nakade, J.; Kita, K.; Nakagawa, T.; Nanjo, S.; Nakamura, T.; Matsumoto, K.; Soda, M.; Mano, H.; et al. Paracrine receptor activation by microenvironment triggers bypass survival signals and alk inhibitor resistance in eml4-alk lung cancer cells. Clin. Cancer Res. 2012, 18, 3592–3602.
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with alk gene rearranged non–small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482.
- Katayama, R.; Sakashita, T.; Yanagitani, N.; Ninomiya, H.; Horiike, A.; Friboulet, L.; Gainor, J.F.; Motoi, N.; Dobashi, A.; Sakata, S.; et al. P-glycoprotein Mediates Ceritinib Resistance in Anaplastic Lymphoma Kinase-rearranged Non-small Cell Lung Cancer. EBioMedicine 2016, 3, 54–66.
- Costa, D.B.; Shaw, A.T.; Ou, S.-H.I.; Solomon, B.J.; Riely, G.J.; Ahn, M.-J.; Zhou, C.; Shreeve, S.M.; Selaru, P.; Polli, A.; et al. Clinical Experience With Crizotinib in Patients With Advanced ALK-Rearranged Non–Small-Cell Lung Cancer and Brain Metastases. J. Clin. Oncol. 2015, 33, 1881–1888.
- Friboulet, L.; Li, N.; Katayama, R.; Lee, C.C.; Gainor, J.F.; Crystal, A.S.; Michellys, P.-Y.; Awad, M.M.; Yanagitani, N.; Kim, S.; et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non–small cell lung cancer. Cancer Discov. 2014, 4, 662–673.
- Zou, H.Y.; Friboulet, L.; Kodack, D.P.; Engstrom, L.D.; Li, Q.; West, M.; Tang, R.W.; Wang, H.; Tsaparikos, K.; Wang, J.; et al. PF-06463922, an ALK/ROS1 Inhibitor, Overcomes Resistance to First and Second Generation ALK Inhibitors in Preclinical Models. Cancer Cell 2015, 28, 70–81.
- Zhang, Y.; Kent, J.W.; Olivier, M.; Ali, O.; Cerjak, D.; Broeckel, U.; Abdou, R.M.; Dyer, T.D.; Comuzzie, A.G.; Curran, J.E.; et al. A comprehensive analysis of adiponectin QTLs using SNP association, SNP cis-effects on peripheral blood gene expression and gene expression correlation identified novel metabolic syndrome (MetS) genes with potential role in carcinogenesis and systemic inflammation. BMC Med. Genom. 2013, 6, 14.
- Ignatius Ou, S.H.; Azada, M.; Hsiang, D.J.; Herman, J.M.; Kain, T.S.; Siwak-Tapp, C.; Casey, C.; He, J.; Ali, S.M.; Klempner, S.J.; et al. Next-generation sequencing reveals a Novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J. Thorac. Oncol. 2014, 9, 549–553.
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133.
- Sakamoto, H.; Tsukaguchi, T.; Hiroshima, S.; Kodama, T.; Kobayashi, T.; Fukami, T.A.; Oikawa, N.; Tsukuda, T.; Ishii, N.; Aoki, Y. CH5424802, a selective alk inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011, 19, 679–690.
- Lan, S.; Li, H.; Liu, Y.; Xu, J.; Huang, Z.; Yan, S.; Zhang, Q.; Cheng, Y. A Novel ROS1-FBXL17 Fusion Co-Existing with CD74-ROS1 Fusion May Improve Sensitivity to Crizotinib and Prolong Progression-Free Survival of Patients with Lung Ad-enocarcinoma. OncoTargets Ther. 2020, 13, 11499–11504.

