 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bruce Baguley | + 1685 word(s) | 1685 | 2021-02-18 07:52:36 | | | |
| 2 | Camila Xu | Meta information modification | 1685 | 2021-02-25 11:00:15 | | |
Video Upload Options
Amsacrine, an anticancer drug first synthesised in 1970 by Professor Cain and colleagues, showed excellent preclinical activity and underwent clinical trial in 1978 under the auspices of the US National Cancer Institute, showing activity against acute lymphoblastic leukaemia.
1. Introduction
Cancer chemotherapy has gone through three phases over the last 70 years. The first was dominated by the development of cytotoxic drugs that damaged the genetic makeup of the cancer cell, the second was dominated by drug-specific signalling pathways and the third was associated with enhancing host immune responses to eliminate cancer cells. Despite its promise in precision medicine, targeted therapy is limited by the biology of cancer [1], and cytotoxic therapy has been recognised as important for the generation of host responses [2]. One particular focus of the latter has been the development of DNA-binding anticancer drugs. The elucidation of the structure of double-stranded DNA in 1953 by Watson, Crick and Wilkins [3] presented an early, structurally precise molecular target. It is instructive to retrace the logic behind the development of DNA-binding drugs and how they relate to precision cancer therapy. Studies at the Auckland Cancer Society Research Centre (ACSRC), initiated during the late 1960s under the directorship of Professor Bruce Cain, used molecular modelling to design drugs that could intercalate between adjacent DNA base pairs and disrupt the normal function of DNA, leading to anticancer activity. Synthesis of a series of 9-anilinoacridine derivatives identified compounds with significant activity against the L1210 transplantable murine leukaemia [4]. Professor Cain assembled a multidisciplinary team that eventually included medicinal chemists, molecular and cell biologists, pharmacologists and clinical oncologists. Amsacrine, one of a large number of 9-anilinoacridine derivatives synthesised and tested in this programme, was selected for testing in the US National Cancer Institute’s anticancer drug development programme. Amsacrine advanced to Phase I and Phase II clinical trials, was found to have significant activity against human acute leukaemia [5] and entered into worldwide use.
Over a similar time period, research on antibiotics had also led to the identification of DNA-binding drugs that demonstrated antitumour activity [6]. One of the most important was doxorubicin, an anthracycline antibiotic that was found to have utility against a variety of malignancies. A fascinating question to emerge from this early research was why amsacrine’s antitumour activity was limited to leukaemia while doxorubicin’s activity covered a broader spectrum. Subsequent research at the ACSRC has demonstrated the importance of embracing a number of scientific disciplines, including physics, chemistry, molecular biology, toxicology, pharmacology and immunology, in attempts to answer this question.
2. DNA Topoisomerases IIα and IIβ, the Molecular Targets of Amsacrine
One of the most important advances in our understanding of DNA function has been in the field of topology, where it has been shown that three-dimensional structures can be changed without alteration of the nucleotide sequence. DNA topology can be changed by enzymes known as topoisomerases, namely topoisomerase I, which changes the linking number (the number of times a linear polymer is twisted around itself) following reversible breakage of a single strand of the DNA double helix, and topoisomerases IIα and IIβ, which change the linking number following reversible breakage of both DNA strands. Topoisomerases IIα and IIβ have the striking property of being able to pass one double-stranded segment of DNA through another, an essential process in the maintenance of life. Early studies in several laboratories showed that amsacrine induced DNA damage in cultured cells [7][8], but a breakthrough came in 1984 with the finding that amsacrine acted as a poison for topoisomerase II enzymes [9] by inhibiting the religation of broken DNA strands. Doxorubicin was also found to have a similar action [10], thus linking the function of the two drug families, but these findings did not explain why doxorubicin was much more active against solid tumours than was amsacrine.
Professor Bill Denny joined the ACSRC in 1972 and made major contributions to the development of a large series of amsacrine analogues with the aim of determining the optimal structural features for antitumour activity. The development of assays for DNA-binding affinity and kinetics was complemented by the development of cell culture techniques, allowing multiple regression analyses to be carried out to analyse in vitro and in vivo antitumour activity in terms of lipophilic character, base strength and DNA-binding affinity [11]. The latter was found to be necessary but not sufficient for anticancer activity [12]. Molecular modelling suggested that while the acridine moiety was buried in the DNA double helix, the anilino moiety projected from the minor groove, raising the question of whether it might interact directly with the topoisomerase II enzyme.
3. The Discovery of N-[2-(Dimethylamino)-Ethyl]-Acridine-4-Carboxamide (DACA)
In the early 1980s, the ACSRC strategy for the in vivo testing of new DNA-binding drugs shifted from mouse leukaemias (L1210 and P388) to a mouse carcinoma [13], while still paying attention to DNA-binding affinity and physicochemical characteristics. Initially, the Lewis lung (3LL) tumour was used to screen for active drugs, and a large number of new analogues of amsacrine were synthesised and tested. One feature of these results was that some analogues containing a substituted carboxamide substituent on the acridine chromophore showed moderate activity against this lung carcinoma [14]. One derivative, asulacrine, was advanced to Phase I/II clinical trials but showed only modest activity against clinical carcinomas [15]. As synthesis of new analogues in the amsacrine series was being extended, Bruce Cain considered the question of whether the anilino side chain of amsacrine was absolutely required for antitumour activity, and he made the surprising finding that an amsacrine analogue that completely lacked an anilino side chain but contained an amino group on the 9-position and a dimethylaminoethylcarboxamide substituent on the 4-position of the acridine chromophore showed activity against the mouse leukaemia model [16].
Bill Denny took this concept further by considering the question of whether the 9-amino group that linked the anilino side chain to the acridine chromophore was necessary for activity, and he prepared a new compound, DACA, that lacked this group (Figure 1). DACA had only low activity against the P388 leukaemia but surprisingly, at the optimal dose, induced a 100% cure rate of mice carrying the Lewis lung tumour [17]. In structural terms, DACA resembled the anthracycline derivative doxorubicin in having a DNA-intercalating chromophore linked to a positively charged side chain, thus providing a potential link between the acridine and anthracycline series. DACA retained activity as a topoisomerase II poison, but this activity was weaker than that of amsacrine and was accompanied by inhibitory activity against topoisomerases I and II [18]. However, just as the anilino side chain of amsacrine was important for activity against the experimental leukaemia, the dimethylaminoethylcarboxamide side chain of DACA was important for activity against the experimental carcinoma; placement of this side chain at any other position of the acridine chromophore led to inactive compounds [17]. Important clues to the importance of this placement were provided by the results of studies by Wakelin, Denny and others, who showed that the dynamics of dissociation of DACA from the DNA-binding complex were strongly affected by the placement of the side chain [19][20].
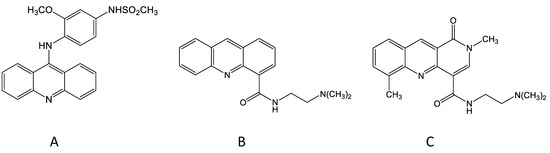
Figure 1. Chemical structures of amsacrine (A), N-[2-(dimethylamino)-ethyl]-acridine-4-carboxamide (DACA) (B) and SN 28049 (C).
4. Toxicological Studies with DACA
The activity of DACA against experimental murine solid tumours, together with evidence of activity in a human tumour xenograft [21], led to a Phase I trial of DACA under the auspices of what is now Cancer Research UK [22][23]. Studies during the dose-escalation phase uncovered an unexpected side effect: pain radiating from the intravenous infusion site. This was mild in some patients, but in others it was sufficiently intense to warrant immediate cessation of the infusion. Although unexpected, such behaviour had been recorded in some patients receiving intravenous doses of another DNA-binding drug, an anthrapyrazole derivative [23]. An important clue as to the cause of the toxicity was provided by the results of pharmacokinetic studies of DACA in mice, in which intraperitoneal doses were tolerated and had excellent antitumour activity but intravenous doses led to clonic seizures and death. The high degree of lipophilicity and the planar structure of DACA suggested interaction with lipids in ion channels, and its activity as an inhibitor of the human ether-à-go-go-related gene (hERG) was tested in patch clamp assays. At concentrations encountered in mice following intravenous administration, DACA showed evidence of hERG toxicity [24][25]. The focus of research was then altered to minimise this toxicity, i.e., to design drugs that were more dose potent than DACA as an antitumour agent but less potent than DACA as an ion channel inhibitor.
5. The Development of the DACA Analogue SN 28049
During the mid-1980s, changes were made in the ACSRC in vivo testing system because of animal ethical considerations; intravenous introduction of Lewis lung tumoursuspensions into mice led to the growth of tumours in the lung and to potential animal suffering. Subcutaneous implantation of tumour cell suspensions avoided this but led to ulcerating subcutaneous tumours, which were also ethically unacceptable. It was found that subcutaneous implantation of another murine tumour, the Colon 38 carcinoma (MCA38), was a more humane alternative, allowing growth delays of subcutaneous tumours to be measured using callipers. DACA was active but not curative against this tumour, meaning that it was a good system in which to search for more active analogues. Initially, DACA analogues in which the acridine chromophore was replaced by phenazine, phenylquinoline or other groups were tested [26], but none were more active against Colon 38 tumours than DACA alone. During this time, Bill Denny had initiated a collaboration with Professor Les Deady at Latrobe University in Melbourne to develop new analogues of DACA; chemical synthesis was carried out in Melbourne while the biological evaluation was carried out at the ACSRC. Analogues containing novel chromophores were developed and tested for DNA affinity, in vitro inhibition of tumour cell proliferation and in vivo growth delay of Colon 38 tumours. Most derivatives had lower in vivo activity than DACA, but some showed unexpectedly high activity [27]. One compound in particular, called SN 28049, showed both higher DNA-binding affinity and higher in vivo dose potency than DACA, and importantly showed lower hERG toxicity than DACA [24]. Significantly, SN 28049, along with some of its derivatives, was found to have curative activity against the Colon 38 tumour [27].
References
- Gatenby, R.A.; Brown, J.S. Integrating evolutionary dynamics into cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 675–686.
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73.
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738.
- Cain, B.F.; Atwell, G.J. The experimental antitumour properties of three congeners of the acridylmethanesulphonanilide (AMSA) series. Eur. J. Cancer Clin. Oncol. 1974, 10, 539–549.
- Arlin, Z.A. Current status of amsacrine (AMSA) combination chemotherapy programs in acute leukemia. Cancer Treat. Rep. 1983, 67, 967–970.
- Arcamone, F. Properties of antitumor anthracyclines and new developments in their application: Cain Memorial Award Lecture. Cancer Res. 1985, 45, 5995–5999.
- Kohn, K.W.; Friedman, C.A.; Ewig, A.G.; Iqbal, Z.M. DNA chain growth during replication of asynchronous L1210 cells. Alkaline elution of large DNA segments from cells lysed on filters. Biochemistry 1974, 13, 4134–4139.
- Ralph, R.K. On the mechanism of action of 4'-[(9-acridinyl)-amino] methanesulphon-m-anisidide. Eur. J. Cancer Clin. Oncol. 1980, 16, 595–600.
- Nelson, E.M.; Tewey, K.; Liu, L.F. Mechanism of antitumor drug action: Poisoning of mammalian DNA topoisomerase II on DNA by 4'-(9-acridinylamino)-methanesulfon-m-anisidide. Proc. Natl. Acad. Sci. USA 1984, 81, 1361–1365.
- Liu, L.F.; Rowe, T.C.; Yang, L.; Tewey, K.M.; Chen, G.L. Cleavage of DNA by mammalian DNA topoisomerase II. J. Biol. Chem. 1983, 258, 15365–15370.
- Baguley, B.C.; Cain, B.F. Comparison of the in vivo and in vitro antileukemic activity of monosubstituted derivatives of 4'-(9-acridinylamino)methanesulfon-m- anisidide. Mol. Pharmacol. 1982, 22, 486–492.
- Baguley, B.C.; Finlay, G.J. Derivatives of amsacrine: Determinants required for high activity against Lewis lung carcinoma. J. Natl. Cancer Inst. 1988, 80, 195–199.
- Baguley, B.C.; Kernohan, A.R.; Wilson, W.R. Divergent activity of derivatives of amsacrine (m-AMSA) towards Lewis lung carcinoma and P388 leukaemia in mice. Eur. J. Cancer Clin. Oncol 1983, 19, 1607–1613.
- Denny, W.A.; Atwell, G.J.; Baguley, B.C. Potential antitumor agents. 40. Orally active 4,5-disubstituted derivatives of amsacrine. J. Med. Chem 1984, 27, 363–367.
- Harvey, V.J.; Hardy, J.R.; Smith, S.; Grove, W.; Baguley, B.C. Phase II study of the amsacrine analogue CI-921 (NSC 343499) in non- small cell lung cancer. Eur. J. Cancer 1991, 27, 1617–1620.
- Atwell, G.J.; Cain, B.F.; Baguley, B.C.; Finlay, G.J.; Denny, W.A. Potential antitumor agents. 43. Synthesis and biological activity of dibasic 9-aminoacridine-4-carboxamides, a new class of antitumor agent. J. Med. Chem. 1984, 27, 1481–1485.
- Atwell, G.J.; Rewcastle, G.W.; Baguley, B.C.; Denny, W.A. Potential antitumor agents. 50. In vivo solid-tumor activity of derivatives of N-[2-(dimethylamino)ethyl]acridine-4-carboxamide. J. Med. Chem. 1987, 30, 664–669.
- Bridewell, D.J.; Finlay, G.J.; Baguley, B.C. Mechanism of cytotoxicity of N-[2-(dimethylamino)ethyl] acridine-4- carboxamide and of its 7-chloro derivative: The roles of topoisomerases I and II. Cancer Chemother. Pharmacol. 1999, 43, 302–308.
- Wakelin, L.P.; Atwell, G.J.; Rewcastle, G.W.; Denny, W.A. Relationships between DNA-binding kinetics and biological activity for the 9-aminoacridine-4-carboxamide class of antitumor agents. J. Med. Chem. 1987, 30, 855–861.
- Wakelin, L.P.; Adams, A.; Denny, W.A. Kinetic studies of the binding of acridinecarboxamide topoisomerase poisons to DNA: Implications for mode of binding of ligands with uncharged chromophores. J. Med. Chem. 2002, 45, 894–901.
- Baguley, B.C.; Zhuang, L.; Marshall, E. Experimental solid tumour activity of N-[2-(dimethylamino)ethyl]-acridine-4-carboxamide. Cancer Chemother. Pharmacol. 1995, 36, 244–248.
- McCrystal, M.R.; Evans, B.D.; Harvey, V.J.; Thompson, P.I.; Porter, D.J.; Baguley, B.C. Phase I study of the cytotoxic agent N-[2-(dimethylamino)ethyl]acridine-4-carboxamide. Cancer Chemother. Pharmacol. 1999, 44, 39–44.
- Leopold, W.R.; Nelson, J.M.; Plowman, J.; Jackson, R.C. Anthrapyrazoles, a new class of intercalating agents with high-level, broad spectrum activity against murine tumors. Cancer Res. 1985, 45, 5532–5539.
- Baguley, B.C. The development of new DNA intercalating anti-cancer drugs. In Horizons in Cancer Research; Watanabe, H.S., Ed.; Nova Publishers, New York: 2012; pp. 47–65.
- Chen, Y.Y.; Finlay, G.J.; Kirker, J.A.; Marshall, E.S.; Richardson, E.; Baguley, B.C. In vivo and in vitro assessment of the action of SN 28049, a benzonaphthyridine derivative targeting topoisomerase II, on the murine Colon 38 carcinoma. Invest. New Drugs 2011, 29, 1504–1510.
- Atwell, G.J.; Baguley, B.C.; Denny, W.A. Potential antitumor agents. 57. 2-Phenylquinoline-8-carboxamides as "minimal" DNA-intercalating antitumor agents with in vivo solid tumor activity. J. Med. Chem. 1989, 32, 396–401.
- Deady, L.W.; Rodemann, T.; Zhuang, L.; Baguley, B.C.; Denny, W.A. Synthesis and cytotoxic activity of carboxamide derivatives of benzo[b][1,6]naphthyridines. J. Med. Chem. 2003, 46, 1049–1054.

