 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andre Larochelle | + 5714 word(s) | 5714 | 2021-02-16 04:09:35 | | | |
| 2 | Dean Liu | -2465 word(s) | 3249 | 2021-02-22 07:21:41 | | |
Video Upload Options
Homology-directed gene editing of hematopoietic stem and progenitor cells (HSPCs) is a promising strategy for the treatment of inherited blood disorders, obviating many of the limitations associated with viral vector-mediated gene therapies.
1. Introduction
The modification or insertion of genes was initially proposed in the early 1970s as a curative approach for inherited disorders[1]. Hematopoietic stem cells (HSCs) are preferred targets for genetic therapies owing to their ability to sustain lifelong hematopoiesis, affording the possibility of durably alleviating a range of conditions. Current gene therapy approaches for inherited blood disorders primarily entail the harvest of hematopoietic stem and progenitor cells (HSPCs) from individuals with an underlying genetic defect and their adoptive transfer after genetic modification ex vivo (Figure 1a). Decades of allogeneic HSPC transplantation performed in the clinic provided a roadmap for therapeutic translation of this novel approach. The avoidance of allo-reactive complications and the reduced complexity of conditioning regimens in autologous transplantation of genetically modified HSPCs provide substantial advantages over allogeneic HSPC transplantation options for these disorders.
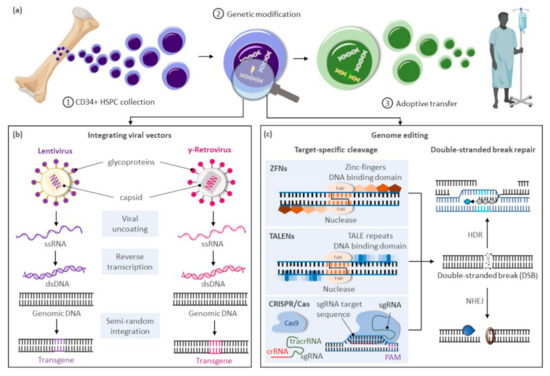
Figure 1. Gene therapy approaches for inherited blood disorders. (a) General scheme for gene therapies of inherited blood disorders: (1) Isolation of CD34+ hematopoietic stem and progenitor cells (HSPCs) from bone marrow harvests or mobilized peripheral blood cell collections; (2) Ex vivo genetic modification of HSPCs; (3) Adoptive transfer of genetically corrected cells to the patient generally following a reduced intensity conditioning regimen to enhance engraftment of the treated cells. (b) Largely random pattern of transgene integration within the target cellular genome after genetic modification of HSPCs using integrating viral vectors based on lentiviruses or gamma-retroviruses. (c) Precise integration of therapeutic genes using genome editing approaches based on zinc-finger nucleases (ZFNs), transcription activator-like effector (TALE) nucleases (TALENS), or the clustered regularly interspaced palindrome repeat (CRISPR)-associated (Cas) platform. Abbreviations: crRNA, CRISPR RNA; dsDNA, double-stranded DNA; DSBs, double-stranded breaks; HDR, homology directed repair; NHEJ, non-homologous end-joining; PAM, protospacer-adjacent motif; sgRNA, single guide RNA; ssRNA, single-stranded RNA; tracrRNA, trans-activating CRISPR RNA.
Clinical trials using gene delivery vectors based on γ-retroviral vectors were initially approved in the 1990s, but only low numbers of corrected cells were detected, and phenotypic correction of the underlying defect was not observed. A refocus on optimizing ex vivo transduction conditions and addition of conditioning regimens to favor engraftment of transduced cells led to the first unequivocal clinical successes in patients with primary immunodeficiencies[2][3][4]. However, the subsequent reporting of malignancies caused by vector-mediated insertional activation of proto-oncogenes in treated patients[5][6][7] encouraged the development of alternative vector designs primarily based on retroviruses of the HIV-1 Lentivirinae subfamily (Figure 1b). Unique constituents of lentiviral vectors facilitate their nuclear translocation within non-dividing HSPCs, further enhancing transduction of these cells. The elimination of portions of the 3′-LTR promoter and enhancer elements in these vectors also provide a key self-inactivating (SIN) safety feature to alleviate concerns on possible recombination with endogenous HIV particles or unintended activation of proto-oncogenes near the genomic site of vector integration. For these SIN vectors, however, the efficiency of transgene expression is highly dependent on the addition of an internal ubiquitous promoter, and reduced or ectopic expression of the therapeutic gene can be limiting in disorders requiring robust or targeted transgene expression for a therapeutic effect.
In recent years, transformative advances have emerged to precisely edit cellular genomes (Figure 1c). Unlike integrating vectors, which can only facilitate gene addition within undefined loci of the cellular genome, novel editing strategies can mediate precise gene correction, gene ablation, and targeted gene addition within cells. Hence, these technologies further address safety concerns associated with integrating vectors, allow more robust and physiologic gene expression by targeted integration of transgenes near endogenous promoters, and extend gene therapies to dominant negative disorders requiring replacement of abnormal gene products rather than simple gene addition. Building on three decades of scientific advances and clinical successes using integrating viral vectors, targeted gene editing in HSPCs is now undergoing similar accelerated pre-clinical and clinical development. In this review, we summarize the current state of targeted gene delivery in HSPCs and examine new strategies developed to improve gene editing efficiency to levels necessary for effective treatment of inherited blood disorders.
2. The Process of Genome Editing
High-efficiency targeted genome editing in mammalian cells generally depends on the initial introduction of a DNA double-stranded break (DSB) at the chosen genomic locus to stimulate cellular DNA repair to yield desired outcomes. As summarized in this section, various cellular nucleases have been engineered to recognize individual target sequences and induce the necessary DSBs and DNA repair response for targeted DNA modification (Figure 1c). Alternative strategies to manipulate cellular genomes that do not rely on double-stranded DNA cleavage, including base editors[8][9][10][11], prime editors[12], and transposases/recombinases [13][14][15][16], were also developed in recent years and have been reviewed elsewhere[17].
2.1. Targeted DNA Double-Stranded Breaks with Engineered DNA Ucleases
Programmable DNA nucleases emerged in the late 1990s and early genome editing studies relied on protein guided synthetic zinc-finger nucleases (ZFNs) (Figure 1c)[18]. ZFNs consist of a non-specific FokI nuclease domain and a finger domain that provides DNA binding specificity. Each amino acid within the finger domain recognizes three DNA base pairs (bp), with several domains required to recognize a 9–18 bp motif. Specific DSBs are made upon dimerization of ZFNs at their FokI domains on opposite strands of the DNA. Zinc-finger nucleases were first shown to successfully edit drosophila DNA in 2002[19] and subsequently in primary human T-cells in 2005[20]. ZFNs have now entered clinical trials[21][22], but their widespread use has been hindered by constraints of the DNA-triplet recognition motif and the specialized expertise required to customize the DNA binding nuclease effector proteins for each genomic target site.
A more versatile transcription activator-like effector (TALE) DNA binding domain from the Xanthomonas spp. proteobacteria[23] was subsequently tethered to the same Fok1 endonuclease domain found in ZFNs to create TALE nucleases (TALENs) (Figure 1c)[24]. TALE domains are modular arrays of conserved repeats of 33–35 amino acids in length. Each repeat binds to a single nucleotide within the target sequence with a binding specificity dictated by the repeat-variable di-residue (RVD) at amino acid positions 12 and 13 of the TALE domain[25]. TALENs have been successfully used in pre-clinical models to edit HSPCs at the CCR5 locus for treatment of HIV[26] and correct the sickle cell mutation in HBB with a single-stranded oligonucleotide (ssODN) donor template[27]. While TALENs’ RVD-DNA recognition code facilitates the design of binding domains with a broader targeting range than ZFNs, TALEN-based gene editing technologies still entail the complex assembly of nucleases specific to each targeted DNA locus.
The bacterial clustered regularly interspaced palindrome repeat (CRISPR) and the CRISPR-associated (Cas) protein, known as CRISPR/Cas, constitutes a novel class of RNA-guided programmable nucleases with unique simplicity and flexibility for targeted gene therapies (Figure 1c)[28]. Identified as a bacterial adaptive immune system[29], CRISPR destroys foreign DNA using the Cas endonuclease in a sequence-specific manner. These naturally occurring immune systems have been categorized as either CRISPR-Cas class 1, which requires complexes composed of several effector proteins for cleavage, or class 2, which allows cleavage of nucleic acids with a single effector domain. Due to their simpler requirements, systems based on class 2 have been favored for genome editing. Class 2 is further partitioned into types II (Cas 9), V (Cas 12), and VI (Cas 13). The type II CRISPR/Cas9 system derived from Streptococcus pyogenes (SpCas9) is currently the most widely used tool for genome editing in hematopoietic and other cellular sources. Cas9 is guided by a dual-RNA complex consisting of a universal trans-activating CRISPR RNA (tracrRNA) that recruits the Cas9 protein, and a CRISPR RNA (crRNA) with homology to a specific DNA sequence. The system was simplified for genome editing applications by synthetic fusion of both RNAs into a single guide RNA (gRNA). Small chemical groups may also be introduced at the extremities of synthesized gRNA to enhance gene editing, as shown at three therapeutically relevant loci in human HSPCs[30]. The Cas9/gRNA ribonucleoprotein (RNP) complex binds to a cognate proto-spacer adjacent motif (PAM) sequence (i.e., NGG) at the target locus, facilitating heteroduplex formation between the guide RNA sequence and the unwound target DNA strand. Cas9 then undergoes conformational changes, which activate its constituent HNH and RuvC nuclease domains to promote cleavage of both target (i.e., bound to the gRNA) and non-target DNA strands, respectively. The process results in formation of predominantly blunt-ended DSBs upstream of the PAM sequence at the chosen locus.
Several Cas9 variants or alternative Cas proteins have been developed to offset limitations of the CRISPR editing system based on SpCas9. For instance, off-target gene editing at unintended sites may result in deleterious cellular effects. Dual-strand targeting using paired Cas9 nickases derived by mutating the RuvC (Cas9D10A) or HNH (H840A) catalytic domains, and two adjacent gRNAs targeting opposing strands of a DNA target[28], can enhance CRIPR/Cas9 accuracy. Similarly, systems based on catalytically inactive Cas9 fused to Fok1 (fCas9), which require recruitment of two Fok1 domains for cleavage[31], can lower the probability of off-target editing. However, design of these systems is more complex, and efficiency is generally lower. Reduced off-target activity was also reported using Cas9 isolated from the alternative bacterial species Streptococcus thermophilus[32] and Francisella novicida (FnCas9)[33], and from type V CRISPR effector Cas12b derived from Bacillus hisashii (BhCas12b)[34]. In HSPCs, the high-fidelity (HiFi) Cas9 mutant improved the on-to-off target ratio when delivered as a purified protein[35], but the potential benefits of other engineered Cas9 variants remain to be determined, as they generally support lower on-target activity[27]. The large cargo size of the CRISPR/SpCas9 system represents another limitation of this technology, precluding packaging within some viral delivery vectors for gene therapy applications. More compact wild-type[36] and mutant[37] Cas9 nucleases from Staphylococcus aureus (SaCas9), Cas9 orthologs derived from Campylobacter jejuni (CjCas9)[38] and Neisseria meningitidis (NmCas9)[39], and type V Cas12e notable for its small size[40] were recently characterized to address this shortcoming. Another disadvantage of the CRISPR/SpCas9 system is the inherent NGG-PAM recognition requirement that limits Cas target site ranges. Several variants have been reported to expand the genome editing armamentarium, such as type V Cas12a nuclease that generally uses orthogonal T-rich PAM sequences[41], NmCas9 that recognizes pyrimidine-rich PAM sequences[39][42], a near PAM-less “SpRY” variant of the prototypical SpCas9[43] and numerous other Cas effectors with altered PAM specificity[44][45].
2.2. Cellular Pathways for Repair of DNA Double-Stranded Breaks
In mammalian cells, DNA DSBs are repaired by classic non-homologous end joining (C-NHEJ), alternative NHEJ (alt-NHEJ, also known as microhomology-mediated end joining, MMEJ), single-strand annealing (SSA), and homology-directed repair (HDR) (Figure 2). The choice of DNA repair pathway after nuclease-mediated DSB formation is influenced by several factors that primarily coalesce on the key role that cell cycle plays in regulating DSB repair[46]. For instance, various phases of the cell cycle will differ in abundance or availability of pathway-specific DNA repair proteins and homologous DNA templates, and the repair mechanism favored may be influenced by the chromatin state of the target cells[47]. In genome editing applications, the DNA end structures induced by distinct programmable nucleases (i.e., blunt ends, 3′ overhangs, or 5′ overhangs) may also trigger distinct cellular pathways for repair of DSBs.
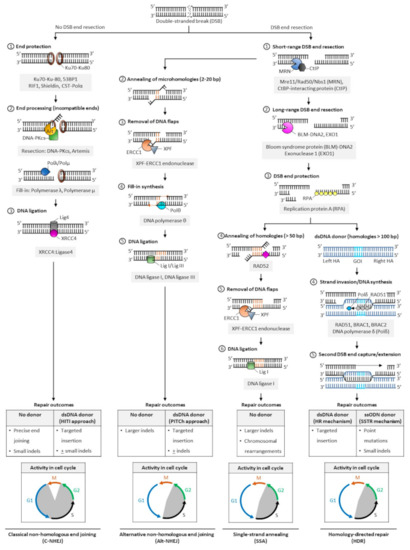
Figure 2. Pathways of DNA double-stranded break repair. Double-stranded breaks (DSBs) are introduced by engineered nucleases at the chosen genomic locus to stimulate endogenous cellular DNA repair mechanisms and promote various repair outcomes. In mammalian cells, DNA DSBs are repaired by classical non-homologous end-joining (C-NHEJ), alternative non-homologous end-joining (alt-NHEJ, also known as MMEJ), single strand annealing (SSA), or homology-directed repair (HDR) pathways. In the absence of donor templates, precise end joining, insertions and deletions (indels), and chromosomal rearrangements may be observed. Addition of a donor DNA template during repair can be used to install and correct point mutations, or knock-in larger DNA sequences. Classic NHEJ does not require template DNA and is the primary repair pathway in cells, whereas alt-NHEJ, SSA, and HDR are known to be active during the S and G2 phases of the cell cycle. Abbreviations: 53BP1: p53-binding protein 1; bp: base pairs; BRAC1: breast cancer type 1; BRAC2: breast cancer type 2; CtBP: C-terminal binding protein; DNA-PKcs: DNA-dependent protein kinase, catalytic subunit; dsDNA: double-stranded DNA; ERCC1: excision repair cross-complementation group 1; GOI: gene of interest; Mre11: meiotic recombination 11; Nbs1: Nijmegen breakage syndrome 1; RAD50/51/52: radiation sensitive 50/51/52; RIF1: Ras relate protein (Rap)-interacting factor 1; CST-Polα: CTC1-STN1-TEN1 (CST)-Polymerase α; HITI: homology-independent targeted insertion; HR: homologous recombination; indels: insertion and deletion; PITCh: precise integration into target chromosome; ssODN: single-strand oligodeoxynucleotide; SSTR: single strand template repair; XPF: xeroderma pigmentosum complementation group F; XRCC4: X-ray repair cross completing protein 4.
The classic form of NHEJ is operational throughout the cell cycle, except mitosis, and quiescent HSPCs largely rely on this mechanism to repair DSB lesions[48][49][50]. Unlike other DSB repair pathways that require DNA end resection at the break site to expose the homology required for repair, broken DNA ends containing no or limited sequence homology (0–4 bp) are ligated in C-NHEJ, and resection is thus not required. Both ends of DSBs are protected from extensive resection by high-affinity binding of Ku70/80 heterodimer complexes[51] and other end protection proteins including the DNA damage response TP53 binding protein 1 (53BP1) and its effectors (RAP1-interacting factor 1 [RIF1], CST complex-polymerase-α [CST-Polα] and the shieldin complex). Compatible DNA ends, such as blunt ends generated by Cas9, are often directly ligated by the XRCC4-DNA ligase 4 (XRCC4:LIG4), with the enhancing activity of XRCC4-like factor (XLF) or paralogue of XRCC4/XLF (PAXX). Incompatible ends, such as 5′/3′-overhangs or 3′-recessed DNA ends, require processing by the Artemis-DNA-PKcs nuclease complex to trim non-complementary end structures, or by Pol µ, Pol λ, and Tdt polymerases to add complementary nucleotides to favor XRCC4:LIG4-mediated ligation. In the absence of donor DNA, the original DNA sequence is generally restored, but limited sequence alterations (e.g., small indels) may also occur at the repair junctions, resulting in silent changes or frameshift mutations leading to target gene inactivation. When a donor sequence is added in vivo along with CRISPR/Cas and gRNA constituents, C-NHEJ can also mediate targeted integration at sites of Cas9-induced DSBs; however, a small percentage of stably integrated sequences may occur in the reverse (undesired) orientation (Figure 2).
The other DSB repair pathways (alt-NHEJ, SSA, and HDR) are known to be active during the S and G2 phases of the cell cycle. They share a first 5′-to-3′ end short-range resection step catalyzed by the Mre11/Rad50/Nbs1 (MRN) endonuclease complex in conjunction with the CtBP-interacting protein (CtIP). This step requires cyclin dependent kinases 1 and 2 (CDK1/2) to phosphorylate and activate CtIP, and is thus limited to the active phases of the cell cycle[52][53]. Recruitment of CtIP to MRN facilitates the removal of Ku70-Ku80 proteins from DSB ends and promotes the dephosphorylation of 53BP1, which in turn inhibits repair by C-NHEJ[54]. This process initially generates 3′ single-stranded overhangs. When short (2–20 bp, most often 3–8 bp) complimentary base pair microhomologies internal to both broken ends are exposed following resection, the broken ends can be repaired by the alt-NHEJ mechanism, involving the annealing of microhomologies, removal of extraneous heterologous DNA flaps by the XPF-ERCC1 endonuclease, fill-in synthesis of the flanking single-stranded regions by DNA Polθ, and sealing by DNA ligases I and III[55][56][57]. Because heterologous flaps flanking the annealed regions of microhomologies are cleaved and lost during alt-NHEJ repair, this pathway is inherently more mutagenic than the classic form of NHEJ. When a donor DNA is added, this repair mechanism can also be exploited for gene knock-in at targeted genomic loci (Figure 2).
In SSA- and HDR-mediated DSB repair, more extensive resection is required. These pathways are thus considerably slower than classical NHEJ or alt-NHEJ mechanisms. The Bloom syndrome protein (BLM)-DNA2 and exonuclease 1 (EXO1) mediate this process, and the replication protein A (RPA) binds the resultant single stranded DNA with high affinity to protect its integrity[58][59]. In SSA, the extended resection exposes longer (> 50 bp) sequences of homologies that are uniquely annealed in a RAD52-dependent manner. Similar to alt-NHEJ, the non-complementary tails are then removed by the XPF-ERCC1 endonuclease complex, and the remaining nicks are sealed by DNA ligase 1[60]. In a normal cellular context, the SSA repair mechanism results in the obligate deletion of a larger sequence between homologous repeats and may promote chromosomal rearrangements (Figure 2)[60].
In HDR[61], RAD51 recombinase is recruited in an ATP-dependent manner to RPA-coated single-stranded DNA, forming a RAD51-DNA nucleoprotein filament. This process is mediated by BRACA2, which is recruited to DNA DSBs by PALB2 and BRAC1 in humans[62][63][64]. The RAD51 ssDNA filament then locates a homologous DNA template. The template is generally a double-stranded sister chromatid available in late S/G2 phases of the cell cycle[52][65][66], or can be provided exogenously in genome editing applications in the form of a double-stranded donor flanked by homology arms. A homology tract of more than 100 bp is typically required as a template to initiate repair by homologous recombination. When complementary ssODN are used as DNA donors, DSBs are processed by a distinct mechanism, the single-strand template repair (SSTR) pathway, which is independent of RAD51 but requires an operative Fanconi anemia (FA) pathway and at least two RAD51 paralogs (RAD51C and XRCC3)[67]. In HDR, the ssDNA filament then invades the homologous region to form a displacement (D)-loop where the template DNA is copied by DNA polymerase δ (Pol δ). The second DSB end is eventually captured by the invading strand, forming a DNA intermediate with two Holliday junctions. This structure undergoes gap repair DNA filling and ligation, and is ultimately resolved at both Holliday junctions in a non-crossover or crossover mode. In some cases, repair can occur by synthesis dependent strand annealing (SDSA), in which the newly replicated DNA dissociates from the template without the formation of a Holliday junction, or by break-induced replication (BIR), when the second DSB end is absent or cannot be found; however, the role of these pathways in genome editing has not been defined (Figure 2). Owing to the obligate use of a donor template sequence, HDR is considered error-free and is generally the preferred pathway for genome editing. Site- and orientation-specific integration at a chosen locus, either upstream of an endogenous promoter or within a safe harbor locus, is a commonly desired repair outcome for therapeutic applications. However, the low frequency of HDR in primary cells, especially long-term repopulating HSCs, remains a challenge to achieving high rates of targeted gene insertion by HDR[48][68].
2.3. Cellular Delivery of Gene Editing Tools
Safe and effective cellular delivery of engineered nucleases, gRNAs, and template sequences constitutes a key step in the process of gene editing. For ex vivo genome editing, approaches for the delivery of the required constituents within target cells can be broadly classified into viral vectors, electroporation, and cell-penetrating peptides[69]. In primary cells, including HSPCs, nucleases and the associated gRNAs are most effectively delivered by electroporation of mRNA molecules or as an RNP complex between gRNAs and the nuclease (e.g., Cas9) protein. Unlike transfection of DNA plasmid molecules, this approach results in limited cytotoxicity to HSPCs. In addition, the short half-life of the complex temporally limits the nuclease activity and the likelihood of genome editing at off-target loci[70].
Donor template delivery has also been a significant challenge for gene editing of HSPCs, as electroporation of dsDNA is highly toxic. Several alternative delivery platforms have been successfully been used, including ssODNs co-delivered with Cas9 [27][71][72][73]. For larger gene insertions, viral vectors are very effective, including integrase deficient lentivirus (IDLV)[74][75][76] and adeno associated virus serotype 6 (AAV6)[77]. There are several caveats, however, to the delivery of donor templates using virus-based systems, including DNA packaging capacity, which is limited by the viral capsid size, and off-target integration of viral genes[78].
References
- Friedmann, T.; Roblin, R. Gene therapy for human genetic disease? Science 1972, 175, 949–955.
- Cavazzana-Calvo, M.; Hacein-Bey, S.; de Saint Basile, G.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.L.; et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease [see comments]. Science 2000, 288, 669–672.
- Hacein-Bey-Abina, S.; Hauer, J.; Lim, A.; Picard, C.; Wang, G.P.; Berry, C.C.; Martinache, C.; Rieux-Laucat, F.; Latour, S.; Belohradsky, B.H.; et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2010, 363, 355–364.
- Hacein-Bey-Abina, S.; Le Deist, F.; Carlier, F.; Bouneaud, C.; Hue, C.; De Villartay, J.P.; Thrasher, A.J.; Wulffraat, N.; Sorensen, R.; Dupuis-Girod, S.; et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N. Engl. J. Med. 2002, 346, 1185–1193.
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142.
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256.
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419.
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471.
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424.
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788.
- Yang, B.; Yang, L.; Chen, J. Development and Application of Base Editors. CRISPR J. 2019, 2, 91–104.
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157.
- Chaikind, B.; Bessen, J.L.; Thompson, D.B.; Hu, J.H.; Liu, D.R. A programmable Cas9-serine recombinase fusion protein that operates on DNA sequences in mammalian cells. Nucleic Acids Res. 2016, 44, 9758–9770.
- Chen, S.P.; Wang, H.H. An Engineered Cas-Transposon System for Programmable and Site-Directed DNA Transpositions. CRISPR J. 2019, 2, 376–394.
- Klompe, S.E.; Vo, P.L.H.; Halpin-Healy, T.S.; Sternberg, S.H. Transposon-encoded CRISPR-Cas systems direct RNA-guided DNA integration. Nature 2019, 571, 219–225.
- Strecker, J.; Ladha, A.; Gardner, Z.; Schmid-Burgk, J.L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. RNA-guided DNA insertion with CRISPR-associated transposases. Science 2019, 365, 48–53.
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844.
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160.
- Bibikova, M.; Golic, M.; Golic, K.G.; Carroll, D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 2002, 161, 1169–1175.
- Urnov, F.D.; Miller, J.C.; Lee, Y.L.; Beausejour, C.M.; Rock, J.M.; Augustus, S.; Jamieson, A.C.; Porteus, M.H.; Gregory, P.D.; Holmes, M.C. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 2005, 435, 646–651.
- Porteus, M.H. A New Class of Medicines through DNA Editing. N. Engl. J. Med. 2019, 380, 947–959.
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359.
- Boch, J.; Bonas, U. Xanthomonas AvrBs3 family-type III effectors: Discovery and function. Annu. Rev. Phytopathol. 2010, 48, 419–436.
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512.
- Bogdanove, A.J.; Voytas, D.F. TAL effectors: Customizable proteins for DNA targeting. Science 2011, 333, 1843–1846.
- Saydaminova, K.; Ye, X.; Wang, H.; Richter, M.; Ho, M.; Chen, H.; Xu, N.; Kim, J.S.; Papapetrou, E.; Holmes, M.C.; et al. Efficient genome editing in hematopoietic stem cells with helper-dependent Ad5/35 vectors expressing site-specific endonucleases under microRNA regulation. Mol. Ther. Methods Clin. Dev. 2015, 1, 14057.
- DeWitt, M.A.; Magis, W.; Bray, N.L.; Wang, T.; Berman, J.R.; Urbinati, F.; Heo, S.J.; Mitros, T.; Munoz, D.P.; Boffelli, D.; et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016, 8, 360ra134.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821.
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712.
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989.
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 2017, 550, 407–410.
- Muller, M.; Lee, C.M.; Gasiunas, G.; Davis, T.H.; Cradick, T.J.; Siksnys, V.; Bao, G.; Cathomen, T.; Mussolino, C. Streptococcus thermophilus CRISPR-Cas9 Systems Enable Specific Editing of the Human Genome. Mol. Ther. 2016, 24, 636–644.
- Acharya, S.; Mishra, A.; Paul, D.; Ansari, A.H.; Azhar, M.; Kumar, M.; Rauthan, R.; Sharma, N.; Aich, M.; Sinha, D.; et al. Francisella novicida Cas9 interrogates genomic DNA with very high specificity and can be used for mammalian genome editing. Proc. Natl. Acad. Sci. USA 2019, 116, 20959–20968.
- Strecker, J.; Jones, S.; Koopal, B.; Schmid-Burgk, J.; Zetsche, B.; Gao, L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. Engineering of CRISPR-Cas12b for human genome editing. Nat. Commun. 2019, 10, 212.
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018, 24, 1216–1224.
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191.
- Luan, B.; Xu, G.; Feng, M.; Cong, L.; Zhou, R. Combined Computational-Experimental Approach to Explore the Molecular Mechanism of SaCas9 with a Broadened DNA Targeting Range. J. Am. Chem. Soc. 2019, 141, 6545–6552.
- Kim, E.; Koo, T.; Park, S.W.; Kim, D.; Kim, K.; Cho, H.Y.; Song, D.W.; Lee, K.J.; Jung, M.H.; Kim, S.; et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017, 8, 14500.
- Edraki, A.; Mir, A.; Ibraheim, R.; Gainetdinov, I.; Yoon, Y.; Song, C.Q.; Cao, Y.; Gallant, J.; Xue, W.; Rivera-Perez, J.A.; et al. A Compact, High-Accuracy Cas9 with a Dinucleotide PAM for In Vivo Genome Editing. Mol. Cell 2019, 73, 714–726.e4.
- Liu, J.J.; Orlova, N.; Oakes, B.L.; Ma, E.; Spinner, H.B.; Baney, K.L.M.; Chuck, J.; Tan, D.; Knott, G.J.; Harrington, L.B.; et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature 2019, 566, 218–223.
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.; Li, Z.; Peterson, R.T.; Yeh, J.R.; et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485.
- Hou, Z.; Zhang, Y.; Propson, N.E.; Howden, S.E.; Chu, L.F.; Sontheimer, E.J.; Thomson, J.A. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. USA 2013, 110, 15644–15649.
- Walton, R.T.; Christie, K.A.; Whittaker, M.N.; Kleinstiver, B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 2020, 368, 290–296.
- Chatterjee, P.; Jakimo, N.; Lee, J.; Amrani, N.; Rodriguez, T.; Koseki, S.R.T.; Tysinger, E.; Qing, R.; Hao, S.; Sontheimer, E.J.; et al. An engineered ScCas9 with broad PAM range and high specificity and activity. Nat. Biotechnol. 2020, 38, 1154–1158.
- Legut, M.; Daniloski, Z.; Xue, X.; McKenzie, D.; Guo, X.; Wessels, H.H.; Sanjana, N.E. High-Throughput Screens of PAM-Flexible Cas9 Variants for Gene Knockout and Transcriptional Modulation. Cell Rep. 2020, 30, 2859–2868.e5.
- Yeh, C.D.; Richardson, C.D.; Corn, J.E. Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 2019, 21, 1468–1478.
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139.
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; Le Beau, M.M.; Morrison, C.G.; Passegue, E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 2010, 7, 174–185.
- Nijnik, A.; Woodbine, L.; Marchetti, C.; Dawson, S.; Lambe, T.; Liu, C.; Rodrigues, N.P.; Crockford, T.L.; Cabuy, E.; Vindigni, A.; et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature 2007, 447, 686–690.
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729.
- Mimitou, E.P.; Symington, L.S. Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 2010, 29, 3358–3369.
- Ira, G.; Pellicioli, A.; Balijja, A.; Wang, X.; Fiorani, S.; Carotenuto, W.; Liberi, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004, 431, 1011–1017.
- Peterson, S.E.; Li, Y.; Chait, B.T.; Gottesman, M.E.; Baer, R.; Gautier, J. Cdk1 uncouples CtIP-dependent resection and Rad51 filament formation during M-phase double-strand break repair. J. Cell Biol. 2011, 194, 705–720.
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463.
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714.
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725.
- Yan, C.T.; Boboila, C.; Souza, E.K.; Franco, S.; Hickernell, T.R.; Murphy, M.; Gumaste, S.; Geyer, M.; Zarrin, A.A.; Manis, J.P.; et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 2007, 449, 478–482.
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514.
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362.
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575.
- Jasin, M.; de Villiers, J.; Weber, F.; Schaffner, W. High frequency of homologous recombination in mammalian cells between endogenous and introduced SV40 genomes. Cell 1985, 43, 695–703.
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683.
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262.
- Thorslund, T.; McIlwraith, M.J.; Compton, S.A.; Lekomtsev, S.; Petronczki, M.; Griffith, J.D.; West, S.C. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1263–1265.
- Kadyk, L.C.; Hartwell, L.H. Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 1992, 132, 387–402.
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308.
- Richardson, C.D.; Kazane, K.R.; Feng, S.J.; Zelin, E.; Bray, N.L.; Schafer, A.J.; Floor, S.N.; Corn, J.E. CRISPR-Cas9 genome editing in human cells occurs via the Fanconi anemia pathway. Nat. Genet. 2018, 50, 1132–1139.
- Lomova, A.; Clark, D.N.; Campo-Fernandez, B.; Flores-Bjurstrom, C.; Kaufman, M.L.; Fitz-Gibbon, S.; Wang, X.; Miyahira, E.Y.; Brown, D.; DeWitt, M.A.; et al. Improving Gene Editing Outcomes in Human Hematopoietic Stem and Progenitor Cells by Temporal Control of DNA Repair. Stem Cells 2019, 37, 284–294.
- Ates, I.; Rathbone, T.; Stuart, C.; Bridges, P.H.; Cottle, R.N. Delivery Approaches for Therapeutic Genome Editing and Challenges. Genes 2020, 11, 1113.
- Smith, R.H.; Chen, Y.C.; Seifuddin, F.; Hupalo, D.; Alba, C.; Reger, R.; Tian, X.; Araki, D.; Dalgard, C.L.; Childs, R.W.; et al. Genome-Wide Analysis of Off-Target CRISPR/Cas9 Activity in Single-Cell-Derived Human Hematopoietic Stem and Progenitor Cell Clones. Genes 2020, 11, 1501.
- De Ravin, S.S.; Li, L.; Wu, X.; Choi, U.; Allen, C.; Koontz, S.; Lee, J.; Theobald-Whiting, N.; Chu, J.; Garofalo, M.; et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci. Transl. Med. 2017, 9.
- Riesenberg, S.; Chintalapati, M.; Macak, D.; Kanis, P.; Maricic, T.; Paabo, S. Simultaneous precise editing of multiple genes in human cells. Nucleic Acids Res. 2019, 47, e116.
- Wienert, B.; Nguyen, D.N.; Guenther, A.; Feng, S.J.; Locke, M.N.; Wyman, S.K.; Shin, J.; Kazane, K.R.; Gregory, G.L.; Carter, M.A.M.; et al. Timed inhibition of CDC7 increases CRISPR-Cas9 mediated templated repair. Nat. Commun. 2020, 11, 2109.
- Hoban, M.D.; Cost, G.J.; Mendel, M.C.; Romero, Z.; Kaufman, M.L.; Joglekar, A.V.; Ho, M.; Lumaquin, D.; Gray, D.; Lill, G.R.; et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood 2015, 125, 2597–2604.
- Hoban, M.D.; Lumaquin, D.; Kuo, C.Y.; Romero, Z.; Long, J.; Ho, M.; Young, C.S.; Mojadidi, M.; Fitz-Gibbon, S.; Cooper, A.R.; et al. CRISPR/Cas9-Mediated Correction of the Sickle Mutation in Human CD34+ cells. Mol. Ther. 2016, 24, 1561–1569.
- Genovese, P.; Schiroli, G.; Escobar, G.; Tomaso, T.D.; Firrito, C.; Calabria, A.; Moi, D.; Mazzieri, R.; Bonini, C.; Holmes, M.C.; et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature 2014, 510, 235–240.
- Wang, J.; Exline, C.M.; DeClercq, J.J.; Llewellyn, G.N.; Hayward, S.B.; Li, P.W.; Shivak, D.A.; Surosky, R.T.; Gregory, P.D.; Holmes, M.C.; et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 2015, 33, 1256–1263.
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E.; et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019, 10, 4439.


