 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pawel Stankiewicz | + 2352 word(s) | 2352 | 2021-02-03 06:53:48 | | | |
| 2 | Bruce Ren | -55 word(s) | 2297 | 2021-02-20 04:48:48 | | |
Video Upload Options
The FOXF1 Adjacent Noncoding Developmental Regulatory RNA (Fendrr) plays an important role in the control of gene expression in mammals. It is transcribed in the opposite direction to the neighboring Foxf1 gene with which it shares a region containing promoters. In humans, FENDRR is located on chromosome 16q24.1, and is positively regulated both by the FOXF1 distant lung-specific cis-acting enhancer and by trans-acting FOXF1. Fendrr has been shown to function as a competing endogenous RNA, sponging microRNAs and protein factors that control stability of mRNAs, and as an epigenetic modifier of chromatin structure around gene promoters and other regulatory sites, targeting them with histone methyltrasferase complexes. In mice, Fendrr is essential for development of the heart, lungs, and gastrointestinal system; its homozygous loss causes embryonic or perinatal lethality. Importantly, deregulation of FENDRR expression has been causatively linked also to tumorigenesis, resistance to chemotherapy, fibrosis, and inflammatory dis-eases.
1. Introduction
Nearly the entire human genome is transcribed; however, only approximately 2% of the transcriptome becomes translated into polypeptides longer than 100 amino acids [1][2]. Long non-coding RNAs (lncRNAs) are classified as transcripts of more than 200 nt in length, showing a very limited translational potential. Like mRNAs, most lncRNAs are synthesized by RNA polymerase II and can be capped, polyadenylated, or spliced. LncRNAs have been found in the nucleus and/or cytoplasm. They can functionally interact with microRNAs (miRs), mRNAs, dsDNA, or proteins [3][4].
A subset of anti-sense lncRNAs have their 5′ ends located in proximity to the 5′ ends of the protein-coding genes transcribed in the opposite direction. In some cases, those divergent genes may overlap (e.g., RNF157-AS1 and FOXJ1, FOXC2-AS1 and FOXC2, ZCCHC14-DT and ZCCHC14). In other cases, like lncRNA gene FENDRR (FOXF1 Adjacent Noncoding Developmental Regulatory RNA; HGNC: 43894, MIM: 614975) and the transcription factor (TF)-coding FOXF1, they share a genomic region containing promoters. The symbol FENDRR is an acronym for the Fetal-lethal Non-coding Developmental Regulatory RNA and reflects the significance of this lncRNA in early embryonic development [5]. Previously, FENDRR was referred to as FOXF1-AS1, lincFOXF1, onco-lncRNA-21, or TCONS_00024240. FENDRR was identified in the studies that expanded the catalog of human lncRNAs to over 3000 based on their (i) distinctive histone 3 (H3)-based chromatin signature (H3K4me3-H3K36me3) that marks transcribed genes and (ii) limited protein coding potential [6].
2.FENDRR Structure
In humans, FENDRR maps to chromosome 16q24.1 at chr16:86,474,529-86,509,099 (GRCh38; ENSG00000268388, RNAcentral: http://rnacentral.org, [7]) (Figure 1A). However, due to a variety of FENDRR transcription start sites (TSS) and alternative splicing, the combined genomic region coding for all FENDRR transcripts is actually larger, ~37 kb in size, mapping between chr16:86,473,637 for the 3′ end of the FENDRR:25 transcript and chr16:86,510,615 for the 5′ end of the HSALNT0234884 transcript (LncBook: http://bigd.big.ac.cn/lncbook, [8]) (Figure 1A,B). Due to the presence of two to six (10 in the NONHSAT174160.1 isoform) relatively short exons, FENDRR isoforms are only ~0.4–4 kb in size after splicing. LncBook, a curated database of human lncRNAs, incorporated in a non-coding RNA database RNAcentral, lists 50 FENDRR transcripts (examples are shown in Figure 1A,B). Fourteen of them are annotated in the GENCODE database (http://www.gencodegenes.org, [9]) as FENDRR-201 to 214 (Figure 1A). The diversity of FENDRR splicing contrasts with that of the neighboring protein-coding FOXF1 gene that features only a single isoform in humans, and supports a contention [10] that non-coding genes undergo alternative splicing more often than the protein-coding genes.
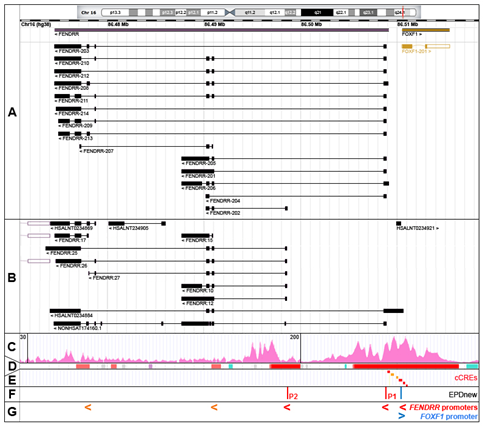
Figure 1. The FOXF1 Adjacent Noncoding Developmental Regulatory RNA (FENDRR) gene, alternatively spliced FENDRR transcripts, and promoters. (A) GENCODE annotated FENDRR isoforms (RNAcentral browser screenshot). (B) Selected additional FENDRR isoforms listed in RNAcentral. (C) Chromatin histone 3 modification, H3K4me3, usually found around active promoters (ENCODE ChIP-seq data from human lung fibroblasts, HLFs). (D) Ensembl-annotated promoters (red) and promoter flanking regions (orange). (E) ENCODE candidate cis-regulatory elements (cCREs, promoters are in red). (F) Eukaryotic promoter database (EPDnew)-annotated promoters. (G) Compilation of FENDRR promoters: EPDnew-annotated, cCRE promoters (red) and cCRE promoter flanking regions (orange). Note that promoter locations correlate with 5′ ends of the majority of FENDRR isoforms.
FENDRR has been predicted to have a very low coding capability based on PRIDE reprocessing 2.0 (=0), PhyloCSF score (=65.5245), CPAT coding probability (=28.39%), and Ribosome-profiling: Lee translation initiation sites (=0) and Bazzini small ORFs (=0) predictions (LNCipedia: http://lncipedia.org). In contrast to evolutionary conservation of FOXF1, the orthologs of human FENDRR are best conserved among higher primates and have not been identified in animals other than mammals (http://genome.ucsc.edu/ENCODE).
3.FENDRR Transcription
3.1. Promoter
The chromatin hallmarks of a human promoter include (i) a nucleosome-free region around and upstream of a transcription start site (TSS), (ii) a peak of RNA polymerase II binding slightly downstream of TSS and usually overlapping with the TF binding sites, and (iii) several H3K4me3-marked nucleosomes especially in the downstream portion of the promoter region [11]. Taking into account these criteria, FENDRR transcription can start from at least three promoters. The majority of the FENDRR isoforms are transcribed from the intergenic promoter P1, annotated in the Eukaryotic promoter database (EPDnew, http://epd.vital-it.ch, [11]) as a promoter element mapping at chr16:86,508,876–86,508,935 and in ENCODE as candidate cis-regulatory element (cCRE) mapping at chr16:86,508,968–86,509,250 (Figure 1C–F). This promoter is located within a large island of 365 CpGs and is sensitive to DNA methylation. It belongs to a class of promoters with a dispersed TSS pattern. The emerging view on the functioning of human promoters is that they are intrinsically bi-directional and their actual directionality is controlled both at the transcriptional and/or post-transcriptional levels [12][13][14][15][16]. In support of this notion, an abortive transcription (the HSALNT0234921 transcript mapping to chr16:86,510,010–86,510,341) originates possibly from this promoter in the direction opposite to the FENDRR transcription (Figure 1B). The FENDRR promoter with the apparently second highest usage, P2 (EPDnew-annotated element: chr16:86,498,542–86,498,601), is located within FENDRR intron 1 and may initiate FENDRR-202, FENDRR:10 to 12, 26, 27, and 29 transcripts (Figure 1). As in the case of the intergenic promoter, the intragenic FENDRR promoter P2 is located within an island of 184 CpGs. It is possible that FENDRR-207, and FENDRR:14 to 17 transcripts initiate from two other intragenic promoters located further downstream (Figure 1). Interestingly, one of the FENDRR isoforms, the HSALNT0234884 transcript (chr16:86,474,121–86,510,614), begins with a cCRE (chr16:86,510,620–86,510,824) overlapping the 5′ end of the non-coding portion of the FOXF1 exon 1 (Figure 1).
3.2. Enhancer
The lung-specific FOXF1 enhancer is located ~270 kb upstream to the FOXF1 gene [17][18][19][20][21]. This enhancer was originally described as ~60 kb-large regulatory region based on the overlap of heterozygous copy number variant (CNV) deletions detected in patients with Alveolar Capillary Dysplasia with Misalignment of Pulmonary Veins (ACDMPV, MIM: 265380) due to FOXF1 haploinsufficiency [17]. It consists of six regulatory elements annotated as enhancers in GeneHancer database [22], each overlapping several cCREs (Figure 2A). In cultured human lung fibroblasts (HLFs) these regulatory elements feature H3K27ac and H3K4me1 chromatin modifications which are the predominant H3 marks at the nucleosomes flanking the active/poised enhancer elements (ENCODE). Moreover, chromosome circular conformation capture (4C) analysis in human pulmonary microvascular endothelial cells and 3C analysis in cells isolated from mouse embryonic lungs showed that this distant enhancer physically interacts with the FENDRR-FOXF1 intergenic promoter region. Interestingly, we have found a ~35 kb-large genomic instability hotspot, featuring the evolutionarily young LINE1 elements, L1PA2 and L1PA3 flanking five Alu repeats, located at the distal edge of this enhancer region, and responsible for several pathogenic enhancer deletions of which the distal breakpoints are mapped within the hotspot [23]. The appearance of this genomic instability hotspot in the course of evolution correlates with the branching out of the Homo-Pan-Gorilla clade.
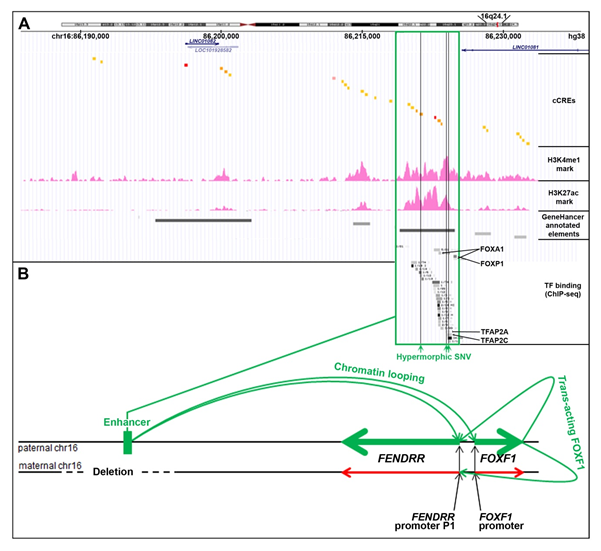
Figure 2. Cis- and trans-regulation of the FENDRR expression in the lungs (modified from [24]). (A) FENDRR-FOXF1 distant enhancer region, located ~250 kb centromerically to the 3′ end of FENDRR (UCSC genome browser screenshot). The most essential part of this enhancer is shown in green frame. The enhancer features histone 3 modifications, usually found in active enhancers, and ChIP-seq-determined in HLFs binding sites for numerous transcription factors (TF): FOXA1, FOXP1, CEBPB, MAFK, RAD21, SMARCC1, CTCF, GTF2F1, KAP1, TBP, JUNs, EP300, STAT3, FOS, TFAP2A, and TFAP2C (ENCODE). Single nucleotide variants (SNVs, vertical green lines) that map to the essential part of the enhancer have been proposed to increase activity of the undeleted allele of the enhancer and mitigate ACDMPV phenotype in patients with heterozygous CNV deletions of the enhancer. (B) Scheme of mono-allelic expression of FENDRR from the paternally inherited chromosome 16 in the presence of a heterozygous CNV deletion of the maternal allele of the FENDRR-FOXF1 enhancer. The drawing shown is not to scale.
Based on the overlap of additional pathogenic CNV deletions causative for ACDMPV and the presence of hypermorphic single nucleotide variants (SNVs) in the undeleted allele of the enhancer that significantly ameliorated the lethal ACDMPV phenotype by increasing FOXF1 expression, this enhancer was narrowed to the ~10 kb-large most essential region. This narrowed interval harbors a GeneHancer-annotated regulatory element GH16J086219 (chr16:86,218,986–86,224,837, overlapping with seven cCREs) that corresponds to one of the super-enhancers proposed by Hnisz et al. [25] based on their analysis of the H3K27ac ChIP-seq data from a spectrum of human cell types including fetal lung fibroblasts IMR-90.
Recently, through a correlation of the parental origins of chromosome 16, bearing the heterozygous CNV deletions of this enhancer with that of the transcribed FENDRR allele, we have found that the FOXF1 enhancer regulates in cis also FENDRR (Figure 2B). This finding may help explain the results of in-depth expression analyses of mouse Fendrr/Foxf1 and other lncRNA/protein-coding divergent gene pairs showing that lncRNAs mimic the expression patterns of their protein-coding neighbors [26][27].
Besides the FENDRR/FOXF1 distant enhancer, there are several other regions upstream or downstream to TSS of FENDRR (within a large GeneHancer-annotated element GH16J086491) that, based on their H3 chromatin signature and eQTLs (expression quantitative trait loci), may potentially function as FENDRR proximal enhancers or modulators of its tissue specificity. For instance, in mice, a genomic region located ~1 kb downstream of Foxf1 functions as Foxf1 enhancer in foregut mesoderm and mesenchyme of developing liver and lungs [28]. It was also shown by 3C, using mouse lung cells, that this regulatory element physically interacts in vivo with Fendrr-Foxf1 intergenic region.
3.3. Regulation of FENDRR by FOXF1
In contrast to the co-regulation of FENDRR and FOXF1 expression by the same cis-acting distant enhancer, the involvement in this regulation of the trans-acting TF FOXF1 was unexpected. Depletion of FOXF1 in lung fibroblasts by siRNA or FOXF1 point mutations causative for ACDMPV were found to correlate with a substantial (~50%) decrease in FENDRR levels (measured by qPCR with TaqMan assay for the exon 1/2 junction, and RNA-seq, respectively). Foxf1 was also shown to likely support Fendrr expression in mice [29]. However, the binding of FOXF1 to the FENDRR promoters or the enhancer has yet to be documented. Interestingly, FENDRR promoters and the enhancer all contain several variants of the FOX TF binding RYAAAYA motif (R = purine, Y = pyrimidine; [30]), suggesting a possibility of direct regulation of FENDRR expression by FOXF1. In support of this notion, ChIP-seq experiments of the TF binding (ENCODE) showed that other members of the FOX TF family, especially FOXA1, bind to the FENDRR major promoter, P1, and the essential region of the enhancer. In addition, FOXF1 might also indirectly regulate FENDRR expression through the control of factors that directly regulate FENDRR. Based on our RNA-seq analyses of the ACDMPV transcriptomes, of about 40 TFs potentially interacting, based on ENCODE’s ChIP-seq data, with the FENDRR primary P1 promoter and the most essential portion of the enhancer, the expression of a histone methyltransferase subunit, ASH2L (binding next to FENDRR P1 promoter’s cCRE), positively correlates with the expression of FOXF1 (reduced by ~40% in ACDMPV cases linked to FOXF1 deficiency). ASH2L can interact with MLL [31] , a Trithorax-group (TrxG) protein involved in histone 3 methylation, H3K4me3, which is usually associated with open chromatin at active promoters. Thus, ASH2L might mediate positive regulation of FENDRR expression by FOXF1.
3.4. Other Factors Controlling FENDRR Transcription
Another potential regulator of FENDRR expression is a pro-apoptotic Annexin 2 (ANXA2) [32]. In electrophoretic mobility shift assay performed using extracts from rat pancreatic acinar cells and fragments of the Fendrr promoter, ANXA2 was specifically bound to the Fendrr intergenic promoter, and the increase in Anxa2 expression in caerulein-treated pancreatic cells positively correlated with the increase in the Fendrr level.
Using a reporter assay in lung cancer cells lines, FENDRR was also shown to be positively regulated by EGR2 and TFAP2A [33]. Both these TFs are known to bind to the FENDRR primary promoter based on ChIP-seq data from lung fibroblasts (ENCODE).
Interestingly, induced expression of Mesp1 during cardiomyocyte differentiation also led to upregulation of several genes including Fendrr [34]. Thus, Fendrr can be a downstream effector of MESP1, a TF best known as a master regulator of cardiovascular system development.
Regarding suppressors of FENDRR, its expression is negatively regulated in the lungs by SMAD3 (but not SMAD2), which is a major signal transducer for cell-membrane Ser/Tyr kinase receptors of TGF-β1 [35]
Of note, using ChIP-seq for TBX2 and TBX4 in IMR-90 fibroblasts, we have found a specific binding of (i) TBX4 at chr16:86,223,833–86,225,160, largely overlapping the GH16J086219 regulatory element in the essential region of the FENDRR-FOXF1 enhancer, (ii) TBX4 at chr16:86,508,364–86,508,853 next to the FENDRR primary promoter, and (iii) TBX2 at 86,507,893–86,509,510, overlapping the FENDRR primary promoter [36]. The functional significance of these interactions is currently unknown, but they suggest the existence of a direct regulatory relationship between TBX4-FGF10 and SHH-FOXF1 signaling pathways during lung development.
Lastly, it has been suggested that FENDRR expression can be regulated through the epigenetic modification of its promoter (P1) that overlaps a large CpG island. The hypermethylation of this promoter has been shown to correlate with suppression of FENDRR expression in gastric cancer-associated fibroblasts [37] Hypermethylation of the FENDRR promoter was also found in 36% of non-small cell lung cancers[38] .
References
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108.
- Mercer, T.R.; Gerhardt, D.J.; Dinger, M.E.; Crawford, J.; Trapnell, C.; Jeddeloh, J.A.; Mattick, J.S.; Rinn, J.L. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat. Biotechnol. 2011, 30, 99–104.
- Mattick, J.S. The state of long non-coding RNA biology. Noncoding RNA 2018, 4, 17.
- Rinn, J.L.; Chang, H.Y. Long noncoding RNAs: Molecular modalities to organismal functions. Annu. Rev. Biochem. 2020, 89, 283–308.
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Währisch, S.; Beisaw, A.; Macura, K.; Bläss, G.; Kellis, M.; Werber, M.; et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell. 2013, 24, 206–214.
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672.
- The RNAcentral Consortium. RNAcentral: A hub of information for non-coding RNA sequences. Nucleic Acids Res. 2019, 47, D221–D229.
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z., Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, 2699.
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774.
- Deveson, I.W.; Brunck, M.E.; Blackburn, J.; Tseng, E.; Hon, T.; Clark, T.A.; Clark, M.B.; Crawford, J.; Dinger, M.E.; Nielsen, L.K.; et al. Universal alternative splicing of noncoding exons. Cell Syst. 2018, 6, 245–255.e5.
- Dreos, R.; Ambrosini, G.; Cavin Périer, R.; Bucher, P. EPD and EPDnew, high-quality promoter resources in the next-generation sequencing era. Nucleic Acids Res. 2013, 41, D157–D164.
- Andersson, R.; Chen, Y.; Core, L.; Lis, J.T.; Sandelin, A.; Jensen, T.H. Human gene promoters are intrinsically bidirectional. Mol. Cell. 2015, 60, 346–347.
- Boque-Sastre, R.; Soler, M.; Oliveira-Mateos, C.; Portela, A.; Moutinho, C.; Sayols, S.; Villanueva, A.; Esteller, M.; Guil, S. Head-to-head antisense transcription and R-loop formation promotes transcriptional activation. Proc. Natl. Acad. Sci. USA 2015, 112, 5785–5790.
- Lacadie, S.A.; Ibrahim, M.M.; Gokhale, S.A.; Ohler, U. Divergent transcription and epigenetic directionality of human pro-moters. FEBS J. 2016, 283, 4214–4222.
- Jin, Y.; Eser, U.; Struhl, K.; Churchman, L.S. The ground state and evolution of promoter region directionality. Cell 2017, 170, 889–898.
- Chiu, A.C.; Suzuki, H.I.; Wu, X.; Mahat, D.B.; Kriz, A.J.; Sharp, P.A. Transcriptional pause sites delineate stable nucleo-some-associated premature polyadenylation suppressed by U1 snRNP. Mol. Cell 2018, 69, 648–663.
- Szafranski, P.; Dharmadhikari, A.V.; Brosens, E.; Gurha, P.; Kolodziejska, K.E.; Zhishuo, O.; Dittwald, P.; Majewski, T.; Mo-han, K.N.; Chen, B.; et al. Small noncoding differentially methylated copy-number variants, including lncRNA genes, cause a lethal lung developmental disorder. Genome Res. 2013, 23, 23–33.
- Dello Russo, P.; Franzoni, A.; Baldan, F.; Puppin, C.; De Maglio, G.; Pittini, C.; Cattarossi, L.; Pizzolitto, S.; Damante, G. A 16q deletion involving FOXF1 enhancer is associated to pulmonary capillary hemangiomatosis. BMC Med. Genet. 2015, 16, 94.
- Seo, H.; Kim, J.; Park, G.H.; Kim, Y.; Cho, S.W. Long-range enhancers modulate Foxf1 transcription in blood vessels of pul-monary vascular network. Histochem. Cell Biol. 2016, 146, 289–300.
- Szafranski, P.; Herrera, C.; Proe, L.A.; Coffman, B.; Kearney, D.L.; Popek, E.; Stankiewicz, P. Narrowing the FOXF1 distant enhancer region on 16q24.1 critical for ACDMPV. Clin. Epigenetics 2016, 8, 112.
- Szafranski, P.; Liu, Q.; Karolak, J.A.; Song, X.; de Leeuw, N.; Faas, B.; Gerychova, R.; Janku, P.; Jezova, M.; Valaskova, I.; et al. Association of rare non-coding SNVs in the lung-specific FOXF1 enhancer with a mitigation of the lethal ACDMPV phe-notype. Hum. Genet. 2019, 138, 1301–1311.
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017, 2017, bax028.
- Szafranski, P.; Kośmider, E.; Liu, Q.; Karolak, J.A.; Currie, L.; Parkash, S.; Kahler, S.G.; Roeder, E.; Littlejohn, R.O.; DeNapo-li, T.S.; et al. LINE- and Alu-containing genomic instability hotspot at 16q24.1 associated with recurrent and nonrecurrent CNV deletions causative for ACDMPV. Hum. Mutat. 2018, 39, 1916–1925.
- Szafranski, P.; Gambin, T.; Karolak, J.A.; Popek, E.; Stankiewicz, P. Unraveling regulation of the FOXF1 adjacent long non-coding RNA FENDRR in lungs. 2021, submitted.
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947.
- Herriges, M.J.; Swarr, D.T.; Morley, M.P.; Rathi, K.S.; Peng, T.; Stewart, K.M.; Morrisey, E.E. Long noncoding RNAs are spa-tially correlated with transcription factors and regulate lung development. Genes Dev. 2014, 28, 1363–1379.
- Cabanski, C.R.; White, N.M.; Dang, H.X.; Silva-Fisher, J.M.; Rauck, C.E.; Cicka, D.; Maher, C.A. Pan-cancer transcriptome analysis reveals long noncoding RNAs with conserved function. RNA Biol. 2015, 12, 628–642.
- Kim, I.M.; Zhou, Y.; Ramakrishna, S.; Hughes, D.E.; Solway, J.; Costa, R.H.; Kalinichenko, V.V. Functional characterization of evolutionarily conserved DNA regions in forkhead box f1 gene locus. J. Biol. Chem. 2005, 280, 37908–37916.
- Ren, X.; Ustiyan, V.; Pradhan, A.; Cai, Y.; Havrilak, J.A.; Bolte, C.S.; Shannon, J.M.; Kalin, T.V.; Kalinichenko, V.V. FOXF1 transcription factor is required for formation of embryonic vasculature by regulating VEGF signaling in endothelial cells. Circ. Res. 2014, 115, 709–720.
- Chen, X.; Ji, Z.; Webber, A.; Sharrocks, A.D. Genome-wide binding studies reveal DNA binding specificity mechanisms and functional interplay amongst Forkhead transcription factors. Nucleic Acids Res. 2016, 44, 1566–1578.
- Yokoyama, A.; Wang, Z.; Wysocka, J.; Sanyal, M.; Aufiero, D.J.; Kitabayashi, I.; Herr, W.;. Cleary, M.L. Leukemia pro-to-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol. Cell Biol. 2004, 24, 5639–5649.
- Zhao, D.; Ge, H.; Ma, B.; Xue, D.; Zhang, W.; Li, Z.; Sun, H. The interaction between ANXA2 and lncRNA Fendrr promotes cell apoptosis in caerulein-induced acute pancreatitis. J. Cell. Biochem. 2018, doi:10.1002/jcb.28097. PMID: 30474876.
- Yang, L.; Wu, D.; Chen, J.; Chen, J.; Qiu, F.; Li, Y.; Liu, L.; Cao, Y.; Yang, B.; Zhou, Y.; et al. A functional CNVR_3425.1 damping lincRNA FENDRR increases lifetime risk of lung cancer and COPD in Chinese. Carcinogenesis 2018, 39, 347–359.
- Scheuermann, J.C.; Boyer, L.A. Getting to the heart of the matter: Long non-coding RNAs in cardiac development and dis-ease. EMBO J. 2013, 32, 1805–1816.
- Huang, C.; Liang, Y.; Zeng, X.; Yang, X.; Xu, D.; Gou, X.; Sathiaseelan, R.; Senavirathna, L.K.; Wang, P.; Liu, L. Long Noncoding RNA FENDRR exhibits antifibrotic activity in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 440–453.
- Karolak, J.A.; Gambin, T.; Szafranski, P.; Stankiewicz, P. Potential interactions between the TBX4-FGF10 and SHH-FOXF1 signaling during human lung development revealed using ChIP-seq. Respiratory Res. 2021, 22, 26.
- Najgebauer, H.; Liloglou, T.; Jithesh, P.V.; Giger, O.T.; Varro, A.; Sanderson, C.M. Integrated omics profiling reveals novel patterns of epigenetic programming in cancer-associated myofibroblasts. Carcinogenesis 2019, 40, 500–512.
- Acha-Sagredo, A.; Uko, B.; Pantazi, P.; Bediaga, N.G.; Moschandrea, C.; Rainbow, L.; Marcus, M.W.; Davies, M.P.A.; Field, J.K.; Liloglou, T. Long non-coding RNA dysregulation is a frequent event in non-small cell lung carcinoma pathogenesis. Br. J. Cancer. 2020, 122, 1050–1058.

